Heterogeneous Catalysts
Здесь есть возможность читать онлайн «Heterogeneous Catalysts» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Heterogeneous Catalysts
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Heterogeneous Catalysts: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Heterogeneous Catalysts»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
tate-of-the-art knowledge of heterogeneous catalysts including new applications in energy and environmental fields
This book focuses on emerging techniques in heterogeneous catalysis, from new methodology for catalysts design and synthesis, surface studies and operando spectroscopies, ab initio techniques, to critical catalytic systems as relevant to energy and the environment. It provides the vision of addressing the foreseeable knowledge gap unfilled by classical knowledge in the field.
Heterogeneous Catalysts: Advanced Design, Characterization and Applications
Presents recent developments in heterogeneous catalysis with emphasis on new fundamentals and emerging techniques Offers a comprehensive look at the important aspects of heterogeneous catalysis Provides an applications-oriented, bottoms-up approach to a high-interest subject that plays a vital role in industry and is widely applied in areas related to energy and environment
is an important book for catalytic chemists, materials scientists, surface chemists, physical chemists, inorganic chemists, chemical engineers, and other professionals working in the chemical industry.
Heterogeneous Catalysts — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Heterogeneous Catalysts», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
6.2 Concept and Advantages of SACs
6.2.1 Concept of SACs
Thomas defined a new class of catalysts as uniform heterogeneous catalysts in 1988 [19]. His group synthesized a Ti‐based single‐site heterogeneous catalyst (SSHC) by grafting metallocene complexes onto mesoporous silica [20]. Heiz and coworkers loaded size‐selected Pd nclusters on MgO(100) films by mass‐selected soft‐landing techniques [21]. Interestingly, they found that a single Pd atom is enough for the production of benzene from acetylene cyclotrimerization. Thomas et al. renamed this class of catalysts as SSHCs [22]. Thomas also categorized SSHCs into four subclasses, one of which includes individual isolated atoms anchored to supports. Böhme and Schwarz proposed the concept of single‐site catalysis in gas‐phase experiments [23]. Qiao et al. observed single Pt atoms anchored on FeO xsurfaces by using high‐resolution high‐angle annular dark‐field‐scanning transmission electron microscopy (HAADF‐STEM), and they coined a new concept of single‐atom catalysis in 2011 [16], thus provoking a hot debate on whether SAs alone can act as active sites in heterogeneous catalysis. Yang et al. generalized the concept and examples of “single‐atom catalysts” in 2013 [17]. Since then, the research on SACs has progressed rapidly. SACs have attracted much attention due to the following aspects.
6.2.2 Advantages of SACs
6.2.2.1 Maximum Atom Efficiency
Noble metals, widely used as catalyst components, are expensive and of limited supply. Thus, enormous efforts have been devoted to reducing the consumption of noble metals. In principle, the CASs of supported noble metal catalysts are either the perimeter atoms of metal NPs in contact with supports or exposed surface atoms of metal NPs [13–15], whereas the metal atoms inside NPs are not involved in catalysis. Thus, constructing SACs is effective for making full use of metal atoms.
6.2.2.2 Unique Catalytic Properties
SACs have been studied in catalytic oxidation, water–gas shift (WGS), hydrogenation, and electrocatalysis, showing superior catalytic performance vs. their counterparts (i.e. supported NPs) [15, 16, 24–26]. The high activity of SACs may be ascribed to the unique coordination of SAs with neighboring atoms of the support as well as metal–support interactions. For example, Pt 1/FeO xSAC showed much higher activity for CO oxidation than Pt NPs supported on FeO x, owing to the partially vacant 5d orbitals of the positively charged, high‐valent Pt atoms [16]. Yang and coworkers reported the use of M 1/TiO 2(M = Pt, Pd, Rh, or Ru) in photocatalytic hydrogen evolution and illustrated a 6‐ to 13‐fold increase in photocatalytic activity compared with the metal clusters loaded onto TiO 2[24]. Furthermore, the active single‐atom sites are well defined, and the identical geometric structure of each active site may result in excellent selectivity compared with the nanoscale counterparts that often have multiple types of active sites. Yan et al. reported that atomically dispersed Pd on graphene showed 100% selectivity to butenes in catalytic hydrogenation of 1,3‐butadiene. In particular, the selectivity to 1‐butene can reach ∼70% at 95% conversion at 50 °C, as explained by the change of 1,3‐butadiene's adsorption mode due to the geometric effect ( Figure 6.2) [25]. Anderson and coworkers reported a study of oxygen reduction reaction catalyzed by size‐selected Pt nclusters deposited on indium tin oxide [26]. The materials showed increased H 2O 2selectivity as the Pt ncluster size decreased, and a maximized H 2O 2selectivity was observed with the smallest Pt 1species [26].
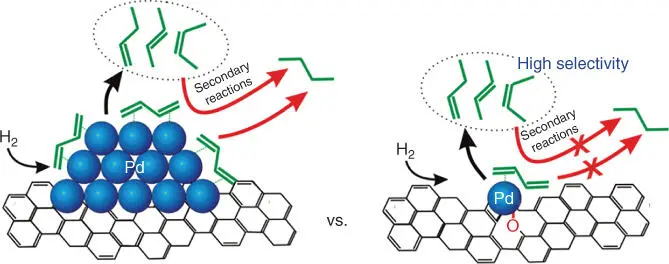
Figure 6.2 Schematic illustration of improvement of selectivity to butenes on single‐atom Pd 1/graphene catalyst.
Source: Yan et al. 2015 [25]. Reproduced with permission of American Chemical Society.
(See online version for color figure).
6.2.2.3 Identification of Catalytically Active Sites
A thorough understanding of the nature of CASs is helpful for improving existing catalysts and designing superior new catalysts [27, 28]. However, the precise identification of CASs of supported metal NP catalysts is challenging. Fujitani and Nakamura found that the CASs of Au/TiO 2for CO oxidation are temperature dependent [29]. At low reaction temperatures the CASs are located at the perimeter interfaces of the Au NPs in contact with TiO 2support, whereas at high temperatures all the surface Au atoms can act as CASs. Ertl's group reported that active metal surfaces are often oscillating during catalytic oxidation [30], meaning that CASs can be changeable.
On the contrary, it is straightforward to identify the CASs of SACs because the single metal atoms in contact with their immediate neighboring atoms of the support surfaces are usually CASs. For example, Yang et al. [31] reported that at higher gold loadings, both gold atoms and NPs existed on titania. After leaching gold NPs by a sodium cyanide solution, the atomically dispersed gold still bound on titania and the catalytic activity in the WGS reaction was intact. Hence, the atomically dispersed gold species with surrounding surface −OH groups should be the CASs in this case. Similar results were reported in other reactions [32, 33]. Therefore, it is easier to study the nature of the CASs by comparing different SACs. We synthesized two thermally stable SACs by two methods, and single Ag atoms were found to be anchored at the {001} top facets of hollandite‐type manganese oxide (HMO) ( Figure 6.3a–d). One SAC denoted as Ag AOR‐HMO was prepared, starting from a supported Ag (particle) sample, by thermal diffusion [34]. The other SAC denoted as Ag IMP‐HMO was prepared by wet impregnation with AgNO 3as a precursor. Thus we can establish the correlation between the electronic structure of CASs and activity by studying the structure and catalytic performance of two single‐atom silver catalysts. Our results showed that the higher depletion of the 4d electronic states of the Ag atoms caused stronger electronic metal–support interactions, leading to easier reducibility of the sample and higher catalytic activity for formaldehyde (HCHO) oxidation [34]. In another work, single‐atom sodium and silver catalysts were also used to differentiate the function of alkalis from that of noble metals under identical conditions [35]. Wang et al. also established a quantitative correlation between the catalytic performance (in NH 3BH 3hydrolysis to generate H 2) and metal–support interactions by using Rh 1/VO 2catalysts [36].
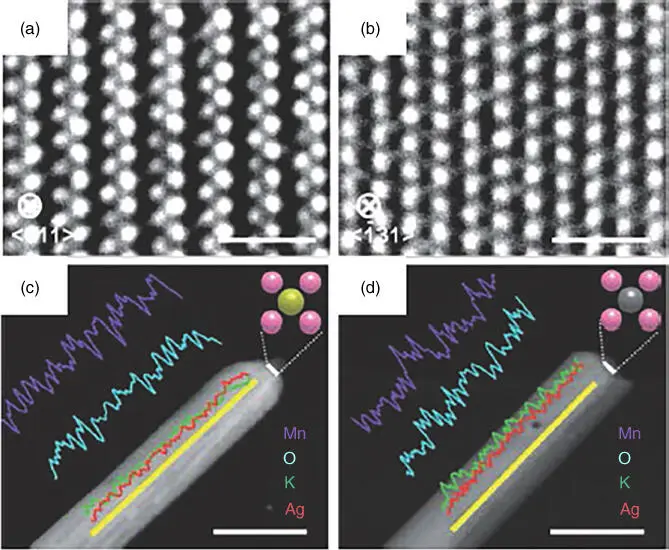
Figure 6.3 High‐resolution TEM and energy dispersive X‐ray (EDX) line scans along the yellow lines of Ag AOR‐HMO (a, c) and Ag IMP‐HMO (b, d). In the structural models, an oxygen atom is represented by a pink ball (c, d), and a silver atom is represented by a yellow ball (c) or a gray ball (d). Scale bars: 1 nm (a, b), 40 nm (c, d).
Source: Hu et al. 2014 [34]. Reproduced with permission of John Wiley & Sons.
(See online version for color figure).
6.2.2.4 Establishment of Intrinsic Reaction Mechanisms
For supported metal NPs, it is challenging to establish an intrinsic reaction mechanism even for a simple model reaction such as CO oxidation. Density functional theory (DFT) calculations can help us understand catalytic reaction mechanisms [16, 37–42]. Because single metal atoms act as the CASs of SACs, establishing an intrinsic reaction mechanism involving single metal atoms thus becomes simplified significantly. For example, the mechanisms of CO oxidation on Ir 1/FeO xor Pt 1/FeO xwere proposed based on DFT calculations and experimental results [16, 39]. The differences in the reaction rates between Ir 1/FeO xand Pt 1/FeO xfor CO oxidation were understood with the help of theoretical investigation. The mechanisms of other reactions such as oxygen reduction reaction [41] and benzene oxidation [42] over SACs were also studied by DFT calculations.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Heterogeneous Catalysts»
Представляем Вашему вниманию похожие книги на «Heterogeneous Catalysts» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.

Обсуждение, отзывы о книге «Heterogeneous Catalysts» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.