Yu Lan - Computational Methods in Organometallic Catalysis
Здесь есть возможность читать онлайн «Yu Lan - Computational Methods in Organometallic Catalysis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Computational Methods in Organometallic Catalysis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Computational Methods in Organometallic Catalysis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Computational Methods in Organometallic Catalysis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Computational Methods in Organometallic Catalysis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Computational Methods in Organometallic Catalysis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
2.2.4 The Third Rung in “Jacob's Ladder” of Density Functionals
The third rung in “Jacob's ladder” of density functionals is meta‐GGA functionals. The variables with more functionals than GGA are the kinetic energy density or the second derivative of the local spin density. The most common meta‐GGA involved are M06‐L, TPSS, and VSXC [45–47], which are often used in computational organometallic chemistry currently.
2.2.5 The Fourth Rung in “Jacob's Ladder” of Density Functionals
The fourth rung in “Jacob's ladder” of density functionals is hybrid‐GGA and hybrid‐meta‐GGA. This kind of functionals are the most popular functional in computational chemistry currently, into which HF exchange is introduced. In the field of computational organometallic chemistry, the commonly used hybrid‐GGA functionals involves B3LYP [42, 48], B97 [49], O3LYP [50], PBE0 [51], m PW1PW [52], X3LYP [53], etc.; the commonly used hybrid‐meta‐GGA functionals involves M05, M05‐2X, M06, M06‐HF, M06‐2X, TPSSh, MPW1K, etc. [46, 54–58].
Undoubtedly, B3LYP is the most widely used functional in computational chemistry, which combines exact HF exchange with Becke's gradient‐corrected exchange, the LYP correlation functional, and VWN for the local correlation terms, as the following function:
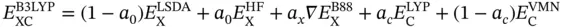
According to Becke's parametrization against the G1 database, a 0, a x, and a care taken as 0.20, 0.72, and 0.81, respectively.
2.2.6 The Fifth Rung in “Jacob's Ladder” of Density Functionals
In the fifth rung of “Jacob's ladder,” the information of virtual orbital is used to build double‐hybrid functional. As examples, in this type, functionals B2PLYP and mPW2PLYP, Kohn–Sham unoccupied orbitals are used to calculate MP2‐type correlation functional [59, 60].
2.2.7 Correction of Dispersion Interaction in Organic Systems
Dispersion interactions are generally described as the interaction between instantaneous dipole moments within the electron distributions of two atoms or molecules. The simplest model of dispersion is the interaction between two Drude oscillators, where the instantaneous dipole moments of the oscillators cause a stabilizing interaction between them.
The dispersion energy, E disp, between two atoms or molecules at large separation takes the form of a series expansion
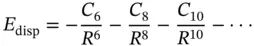
The dispersion coefficients can be determined experimentally or theoretically. Also, there are several approximate methods to evaluate the dispersion coefficients such as the London, Slater–Kirkwood, and Salem models. In most of the cases, dispersion attraction is modeled by the first term in the series only. For example, in molecular mechanics (MM) force fields, commonly used in biochemistry, a Lennard–Jones 6–12 potential is used to describe nonbonded interactions with the R 6term accounting for dispersion attraction and the R 12term corresponding to Pauli repulsion.
Most of the classical density functionals (e.g. B3LYP) cannot describe the dispersion interaction because the long‐range behavior of the correlation functional is not correct. Therefore, the results used to study dispersion‐dominated problems are poor, such as physical adsorption, molecular conformations, ligand coordination, ligand–substrate interaction, and π‐stacking; however, those systems are important in organometallic chemistry. Indeed, classical density functionals reveal poor dispersion interaction is due to the incorrect behavior of the exchange–correlation functional in the medium and long range, especially in the long range, which results in the failure of van der Waals C 6/ R 6behavior.
The frequently used approaches for the dispersion correction of density functionals are grouped in Scheme 2.2, which includes nonlocal van der Waals density functionals (vdW‐DFs), semilocal density functionals parameterizing from the forms of some standard meta‐hybrid‐GGA functionals, DFT‐D methods, and dispersion‐correcting atom‐centered one‐electron potentials (DACACP) [59, 61–65]. The properties of those groups of methods are accounted for in Table 2.1.
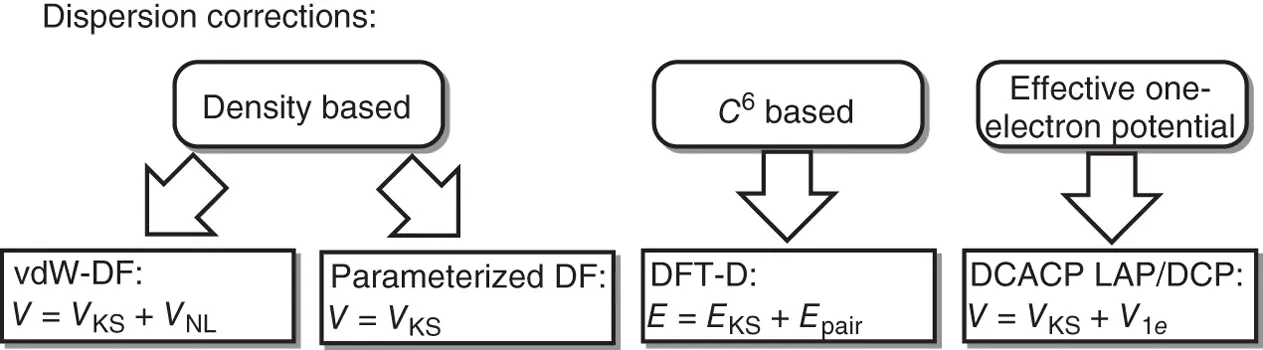
Scheme 2.2 The frequently used approaches for the dispersion correction of density functionals.
Table 2.1 Properties of frequently used approaches for the dispersion correction.
| Properties | vdW‐DF | DF | DFT‐D | DCACP LAP/DCP |
|---|---|---|---|---|
| Correct R −6 | Yes | No | Yes | No |
| Thermochemistry | — | Yes | Yes | — |
| Numerical complexity | High | Medium | Low | Low |
| Simple forces | No | Yes | Yes | Yes |
| System dependency | Yes | Yes | Yes | No |
| Electronic effect | Yes | Yes | No | Yes |
| Empiricism | Low | Medium | Medium | High |
| Analysis insight | — | No | Good | — |
In computational chemistry, some highly parameterized meta‐GGA functionals incorporating kinetic energy density have been assessed to quantitatively account for dispersion effect. One of the most popular parameterized functionals, which can accurately describe dispersion interaction, is Minnesota series of functionals proposed by Truhlar group. Currently, the usually used Minnesota functionals in computational organometallic chemistry include M06, M06‐2X, M06‐L, M11, M11‐L, MN12, MN12‐L, MN15, etc. One serious problem of this and related highly parameterized functionals with many terms in a power series expansion is numerical instability that can lead to artificial van der Waals minima and “noisy” potential energy curves.
DFT‐D methods with semiclassical corrections are alternative choice for computational organometallic chemistry, which treat the difficult dispersion interactions semiclassically and combine the resulting potential with a quantum chemical approach. Currently, the most widely used DFT‐D method, named DFT‐D3 series, was proposed by S. Grimme group in 2006, which represents an update of DFT‐D1. DFT‐D3 has recently been refined regarding higher accuracy, broader range of applicability, and less empiricism [65]. The main new ingredients are atom pairwise‐specific dispersion coefficients and a new set of cutoff radii, both computed from first principles. The coefficients for eighth‐order dispersion terms are computed using established recursion relations. System‐dependent information is used for the first time in a DFT‐D type approach by employing the new concept of fractional coordination numbers. This allows one to distinguish the different hybridization states of atoms in molecules in a differentiable way, which in particular for the first two rows of the periodic table have quite different dispersion coefficients. The method only requires adjustment of two global parameters for each density functional is asymptotically same for a gas of weakly interacting neutral atoms and easily allows the computation of atomic forces. Accurate dispersion coefficients and cutoff radii are available for all elements up to Z = 94. The revised DFT‐D3 method can be used as a general tool for the computation of the dispersion energy in molecules of any kind with DFT and related (low‐cost) electronic structure methods for organometallic systems.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Computational Methods in Organometallic Catalysis»
Представляем Вашему вниманию похожие книги на «Computational Methods in Organometallic Catalysis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.










Обсуждение, отзывы о книге «Computational Methods in Organometallic Catalysis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.