Periodontitis and Systemic Diseases
Здесь есть возможность читать онлайн «Periodontitis and Systemic Diseases» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Periodontitis and Systemic Diseases
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Periodontitis and Systemic Diseases: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Periodontitis and Systemic Diseases»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Periodontitis and Systemic Diseases — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Periodontitis and Systemic Diseases», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Several clinical studies have identified that the relationship between periodontitis and common major systemic pathologies is most probably due to systemic inflammation and bacteraemia.
Indeed, Hasturk and Kantarci 130described two models for the development of systemic inflammation secondary to periodontitis. The first is the dissemination of periodontal pathogens to distant sites via the blood stream (bacteraemia), where they trigger local inflammation. Whilst bacteraemia is a generally accepted occurrence, there is conflicting data on how many bacteria can be found in the systemic circulation in periodontitis or subsequent to mechanical instrumentation.
In the second model, periodontal bacteraemia triggers an acute-phase response by the liver, involving the release of C-reactive protein and production of IL-6, and also activates peripheral blood leukocytes (neutrophils) to release oxygen radicals, thus creating a peripheral oxidative stress response 130. This low-grade peripheral inflammation arising in periodontitis is thought to contribute, in the longer term, to vascular endothelial damage and pancreatic beta-cell damage 59.
1.3.1 Cytokines and inflammatory mediators
In patients with periodontitis, chronic low-level systemic exposure to periodontal microorganisms exists, which leads to significant changes in plasma levels of cytokines and hormones. This systemic response is the link between chronic subclinical inflammation and insulin resistance, initiating the development of T2DM 131. In terms of the inflammatory mediators, there is sufficient evidence that their systemic and local expression is increased in patients with diabetes and periodontitis compared to patients with periodontitis only. For instance, subjects with poorly controlled diabetes and dyslipidaemia have high levels of the eosinophil chemotactic protein eotaxin, macrophage inflammatory protein-1a, granulocyte-macrophage colony-stimulating factor (GM-CSF), TNF-α, IL-6, IL-10 and IL-12 in their GCF 132 , 133. In addition, patients with T2DM have an increased level of lipid peroxidation in the GCF indicated by the detection of malondialdehyde 134. These findings are supported by animal and cell cultures studies 134. Furthermore, studies have shown that a hyperglycaemic state leads to increased expression of innate immunity receptors, such as toll-like receptors (TLR) 2 and 4. Regarding cell function, there is some evidence that diabetes and periodontitis lead to an altered monocyte, T cell and aberrant neutrophil function 135 , 136. Thus, diabetes and hyperglycaemic conditions induce a hyper-inflammatory state systemically and in the infected periodontal tissues, leading to an increase of the disease risk and severity.
1.3.2 Bone homeostasis
Other important evidence is that both T1DM and T2DM modulate alveolar bone homeostasis, which could be an important pathway of periodontal pathogenesis in people with diabetes. In animal studies, rats with diabetes T1DM 137and T2DM 138with experimental ligature- and pathogen-induced periodontitis had a two- to four-fold increase in osteoclast numbers compared with control rats 139. The RANK–RANKL interaction is one of the most potent inducers of osteoclast formation and activity, and osteoprotegerin (OPG) inhibits osteoclast formation binding to RANK like a decoy receptor and thus blocks the activity of RANKL. Animal studies demonstrate that the RANK-RANKL/OPG ratio is a critical factor in the enhanced osteoclastogenesis in periodontitis with diabetes (Figs 1-2 and (1-3) 139 , 140.
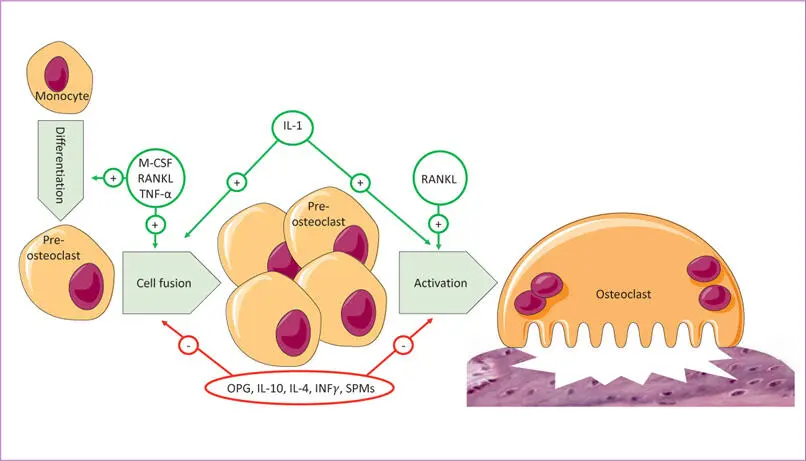
Fig 1-2 Mechanism of osteoclast differentiation and activation (IFNγ = interferon-gamma; IL = interleukin; M-CSF = macrophage colony-stimulating factor; OPG = osteoprotegerin; RANKL = receptor activator of nuclear factor kappa B ligand; SPM = specialised pro-resolving mediators; TNF-α = tumour necrosis factor alpha).
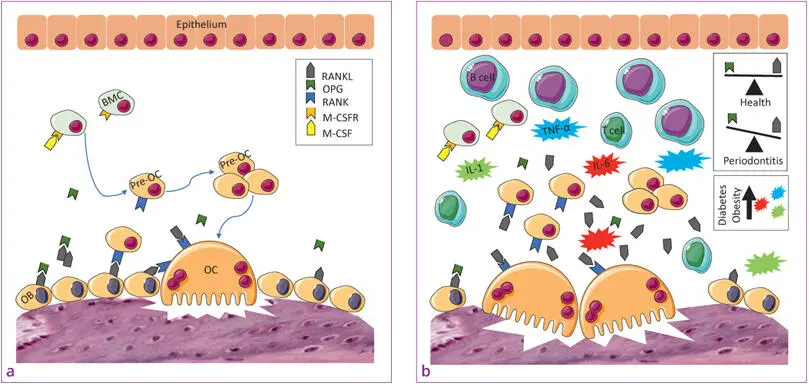
Fig 1-3a and b aOsteoclast differentiation and activity. bPathological bone resorption and the impact of obesity and DM on pro-inflammatory mediator and adipokine accumulation. (BMC = bone marrow cell; IL = interleukin; M-CSF = macrophage colony-stimulating factor; M-CSFR = M-CSF receptor; OB = osteoblast; OC = osteoclast; OPG = osteoprotegerin; RANK = receptor activator of nuclear factor kappa B; RANKL = RANK ligand; TNF-α = tumour necrosis factor alpha.)
1.3.3 Adipokines
Obesity, as a chronic condition, produces a low-grade inflammation, increased chronic oxidative stress, and activation of innate immune system that affects homeostasis over time 141. Several chronic diseases are also the result of obesity (e.g., metabolic syndrome, DM, liver and cardiovascular diseases, and cancer) and associated with oxidative stress.
The obesity-induced pro-inflammatory status affects insulin resistance 142and secretion 143by production and release of adipokines 144. Adipokines are a group of over 600 molecules produced by adipose tissue 145. They act as paracrine and endocrine hormones 146and thus regulate processes like appetite and satiety, fat distribution, inflammation, blood pressure, haemostasis and endothelial function, acting in different organs including adipose tissue itself, brain, liver, muscle and blood vessels 143 , 144. Among the adipokines, adiponectin, leptin, TNF-α, OPG, IL-6, resistin, IL-1, apelin, visfatin, monocyte chemoattractant protein-1 (MCP-1), plasminogen activator inhibitor-1 (PAI-1) and retinol binding protein 4 (RBP4) have been widely investigated 143 , 147. The overproduction of some adipokines in obese subjects is known to contribute to diabetes pathogenesis.
The mechanistic link between obesity, DM and adipose tissue inflammation was first proposed based on the finding that the level of the TNF-α, produced by macrophages, was increased in adipose tissue of obese rodents and humans and that its blockage led to improvement in insulin sensitivity 148. Macrophages are able to infiltrate adipose tissue of obese mice and humans. Nearly 40% to 50% of total cells are macrophages in mice, and the major source of TNF-α 149 , 150. TNF-α binds to TNF receptors 1 and 2, and mediates apoptosis, insulin resistance, lipolysis, inhibition of insulin-stimulated glucose transport and insulin receptor autophosphorylation 146 , 150 , 151. In adipocytes, TNF-α reduces glucose transporter type (GLUT)-4 expression, leading to insulin resistance and atherogenic dyslipidaemia 150.
IL-6 is also released by adipocytes, so it can be considered an adipokine. Obese individuals release greater amounts of IL-6 due to their larger amount and size of adipocytes, explaining a state of low-grade inflammation in these individuals 152 , 153. IL-6 has multiple functions and its exact metabolic role is still controversial. For example, chronically elevated IL-6 levels lead to an impairment of the insulin-mediated glucose uptake by muscle cells 154. On the other hand, acutely elevated IL-6 produced by skeletal muscle during exercise can increase glucose uptake and fatty acid oxidation in these cells 152 , 155. TNF-α and IL-6 are also expressed by inflamed periodontal sites due to microbial stimuli. These mediators enter the systemic circulation, interfere with the function of insulin receptors and thereby derange the process of insulin signalling 156.
Another adipokine, leptin, was the first adipokine known to be associated with direct pancreatic effects and is certainly the most studied of all adipokines. It has a potent inhibitory effect on insulin secretion from pancreatic β-cells, and has the additional effect of reducing pre-pro-insulin gene expression 157; however, these observations are controversial. A study published by Brown et al 158demonstrated that leptin has a U-shape response in human islets, with lower concentrations inhibiting insulin release and higher levels having a relatively stimulatory effect. This finding provides an explanation for the existence of the conflicting reports 158. Patients with periodontitis have reduced leptin levels in their GCF compared with periodontally healthy individuals 159 , 160. On the other hand, periodontitis results in increased plasma levels of leptin, whereas periodontal therapy causes a decrease in the plasma leptin levels 161 , 162. Adiponectin, on the other hand, has beneficial effects on obesity and diabetes. It improves insulin sensitivity and vascular function, thus being both anti-diabetes 163and anti-atherogenic 164. It also inhibits apoptosis of β-cells and increases their proliferation. It is likely that the ratio of adiponectin to leptin (and other adipokines) is a determinant of the effects of the adiposity-induced altered adipokine levels on β-cell function 165.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Periodontitis and Systemic Diseases»
Представляем Вашему вниманию похожие книги на «Periodontitis and Systemic Diseases» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.










![John Bruce - The Lettsomian Lectures on Diseases and Disorders of the Heart and Arteries in Middle and Advanced Life [1900-1901]](/books/749387/john-bruce-the-lettsomian-lectures-on-diseases-and-disorders-of-the-heart-and-arteries-in-middle-and-advanced-life-1900-1901-thumb.webp)

Обсуждение, отзывы о книге «Periodontitis and Systemic Diseases» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.