Prof. Dr. Harald Prof. Dr. Schmidt - Geheilt statt behandelt
Здесь есть возможность читать онлайн «Prof. Dr. Harald Prof. Dr. Schmidt - Geheilt statt behandelt» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на немецком языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Geheilt statt behandelt
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Geheilt statt behandelt: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Geheilt statt behandelt»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Geheilt statt behandelt — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Geheilt statt behandelt», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Der Zusatznutzen eines neuen Medikaments wird in erster Linie durch einen direkten oder geeigneten indirekten Vergleich mit der Standardversorgung anhand der Endpunkte Sterblichkeit, Krankheitshäufigkeit oder gesundheitsbezogene Lebensqualität bestimmt. Der Nachweis erfordert einen statistisch signifikanten Nutzen für patientenrelevante Endpunkte in einer randomisierten (das heißt unter Verwendung eines Zufallsmechanismus besetzten) kontrollierten Studie oder einen sehr großen Nutzen in einer nicht randomisierten Studie.
Wenn ein neu zugelassenes Medikament auf den deutschen Markt kommt, muss die verantwortliche Arzneimittelfirma ein standardisiertes Dossier vorlegen, das alle verfügbaren Belege für den Zusatznutzen des Medikaments gegenüber der Standardversorgung enthält. Nach Markteintritt wird das unabhängige Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) mit der Nutzenbewertung beauftragt. Die Ergebnisse dieser Bewertung dienen als Grundlage für die endgültige Entscheidung, ob ein Zusatznutzen besteht. Dies und sämtliche Stellungnahmen sind auf der Website des Gemeinsamen Bundesausschusses verfügbar. 11
Die Schlussfolgerungen zum Zusatznutzen haben zwei wichtige Funktionen. Erstens dienen sie als Grundlage für Preisverhandlungen zwischen dem Dachverband der gesetzlichen Krankenversicherung und dem Arzneimittelhersteller. Auch wenn der G-BA zu dem Schluss kommt, dass ein neues Arzneimittel keinen Zusatznutzen hat, darf das Arzneimittel auf dem Markt bleiben, darf dann aber nicht mehr als die Standardversorgung kosten. Zweitens können die Schlussfolgerungen für ärztliche Behandlungsleitlinien und individuelle Behandlungsentscheidungen verwendet werden.
2019 veröffentlichte das IQWiG eine Übersicht aller zwischen 2011 und 2017 bewerteten Arzneimittel, die nach der Zulassung auf den deutschen Markt kamen, insgesamt 152 neue Wirkstoffe und 64 bereits zugelassene Arzneimittel in einer neuen Indikation. 12Nur 54 der 216 bewerteten Medikamente (25 Prozent) wurden als Arzneimittel mit einem beträchtlichen oder großen Zusatznutzen eingestuft. Bei 35 (16 Prozent) war der Zusatznutzen entweder gering oder konnte nicht quantifiziert werden. Bei 125 Arzneimitteln (58 Prozent), also bei weit über der Hälfte, konnte kein Zusatznutzen gegenüber der Standardversorgung in Bezug auf Sterblichkeit, Krankheitshäufigkeit oder gesundheitsbezogene Lebensqualität in der zugelassenen Patientenpopulation nachgewiesen werden ( Abbildung 5).
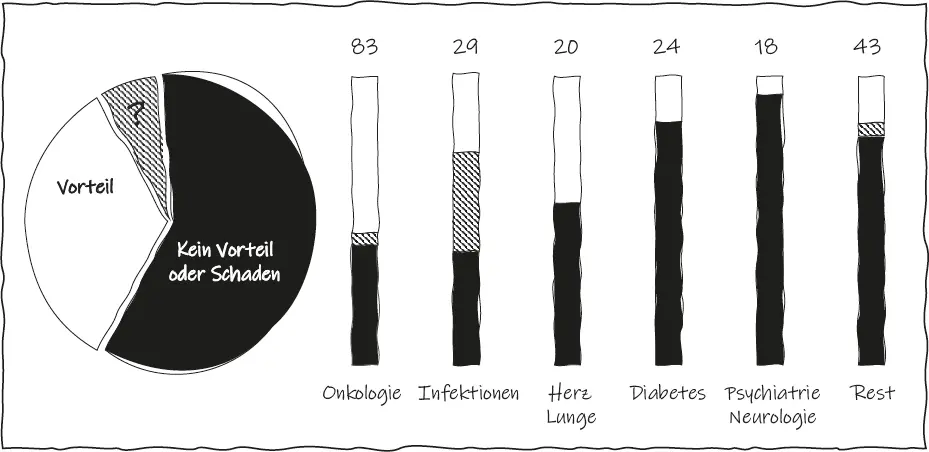
Abb. 5:Die Anteile neu zugelassener Arzneimittel, für die ein zusätzlicher Nutzen gegenüber der bisherigen Standardversorgung besteht (weiß), und solche, bei denen das nicht der Fall ist (schwarz). Insgesamt können also zwei Drittel aller neu zugelassenen Arzneimittel diesen Nachweis nicht erbringen, wobei einige Indikationen wie Psychiatrie / Neurologie und Diabetes besonders schlecht abschneiden.
Schlüsselt man diese Daten noch weiter hinsichtlich der verschiedenen medizinischen Fachgebiete auf, ist die Situation teilweise regelrecht erschreckend. So wurde zum Beispiel in der Psychiatrie/Neurologie und bei Diabetes in nur sechs Prozent (1/18) beziehungsweise 17 Prozent (1/6) der Bewertungen ein Zusatznutzen nachgewiesen (rechter Teil der Abbildung 5). Auch ist zu erkennen, dass die Entwicklung und Zulassung von Arzneimitteln nicht gleichmäßig über die verschiedenen Indikationen verteilt ist; es besteht eine große Neigung der pharmazeutischen Industrie, mehr Krebsmittel und weniger psychiatrische beziehungsweise neurologische Medikamente zu entwickeln. Viele Firmen habe sich aus den letzten beiden Indikationen nahezu zurückgezogen.
Diese Daten sind nicht nur für Deutschland relevant, denn nahezu alle diese Arzneimittel wurden von der Europäischen Arzneimittelagentur (European Medical Agency, EMA) für den Einsatz in ganz Europa zugelassen. Die Ergebnisse spiegeln daher den Stand der gesamten europäischen Arzneimittelentwicklung und -politik wider und verdeutlichen, dass die gesamten Prozesse bis zur Zulassung dringend reformiert werden müssen. Alle Arzneimittelbehörden verfolgen weltweit eine Strategie, die darauf abzielt, die Entwicklung und Zulassung von Medikamenten zu beschleunigen 13, basierend auf der Annahme, ein schnellerer Zugang zu neuen Medikamenten käme den Patienten zugute. Die Rhetorik von Neuheit und Innovation erzeugt den Glauben, dass neue Arzneimittel immer besser seien als bestehende. Zwar gibt es zweifellos dramatische Lücken im Arzneimittelarsenal (siehe die beiden vorherigen Abschnitte „Die meisten Arzneimittel wirken nicht“ und „Die Number Needed to Treat“), aber seit den 1970er-Jahren bietet nur eine begrenzte Anzahl von unter 15 Prozent der neuen Medikamente echte Fortschritte gegenüber den vorhandenen Arzneimitteln und das ohne einen Trend zur Verbesserung. Arzneimittelbehörden wollten den frühen Zugang zu innovativen Medikamenten ermöglichen – oder wurden dazu gedrängt. Die Hoffnung war, das Manko nur begrenzter Informationen zum Zeitpunkt der beschleunigten behördlichen Zulassung dadurch auszugleichen, dass die nachfolgende breite Anwendung am Patienten und hierzu durchgeführte Forschung schließlich den Nutzen für die Patienten belegen würde. 14
Die Realität sieht jedoch ganz anders aus. Beispielsweise zeigte eine systematische Bewertung der Krebsmedikamente, die zwischen 2009 und 2013 von der EMA zugelassen wurden, dass die meisten von ihnen ohne Nachweis eines klinisch sinnvollen Nutzens für patientenrelevante Endpunkte (Überleben und Lebensqualität) zugelassen worden waren und sich einige Jahre später diese Situation kaum verändert hatte. 15Noch beunruhigender ist vielleicht, dass eine systematische Überprüfung neuer Medikamente für über 100 Indikationen, die von der US-Arzneimittelbehörde FDA zugelassen wurden, ergab, dass in weniger als zehn 16beziehungsweise 20 Prozent 17der Fälle eine überlegene Wirksamkeit bestätigt wurde.
Zudem werden solche Post-Marketing-Studien, zu denen die Arzneimittelfirmen nach dem Inverkehrbringen eigentlich verpflichtet sind, häufig nicht durchgeführt und nur zur Hälfte rechtzeitig beziehungsweise innerhalb von fünf bis sechs Jahren abgeschlossen. 18In Deutschland wurde keine der sechs Nachzulassungsstudien, die auf der Grundlage der ersten Bewertung beantragt worden waren und zwischen 2011 und 2017 zur Neubewertung anstehen sollten, tatsächlich durchgeführt. Weltweit tun Aufsichtsbehörden wenig, um nicht kooperierende Unternehmen zu sanktionieren.
Pseudo-Innovation „Me too“
Selbst unter den Medikamenten mit einem Zusatznutzen gibt es viele Pseudo-Innovationen, sogenannte „Me too“-Präparate. „Me too“ heißt im Deutschen „Ich auch“. Hat eine Firma ein wirksames Arzneistoffprinzip entdeckt, ziehen andere Firmen nach und wollen auf Basis desselben Prinzips auch Arzneimittel auf den Markt bringen. Ähnlich wie in der Autoindustrie: Fängt eine Firma an, erfolgreich SUVs zu verkaufen, wollen das alle. Fängt ein anderer an, Mini-SUVs zu verkaufen, ziehen wieder alle nach. Innovation ist das nicht, zumindest würde sich kein Autobauer trauen, das zu behaupten.
So ergab die IQWiG-Analyse, dass in Deutschland 12 von 48 erfolgreichen Bewertungen (25 Prozent) in der Onkologie dasselbe Wirkprinzip hatten. Auch die verschiedenen Medikamente, die bei Hepatitis C einen Zusatznutzen zeigten, verwenden alle einen der drei gleichen Mechanismen oder kombinierten diese. Ein Medikament, das ähnlich ist, bedeutet zwar nicht automatisch, dass es das gleiche ist. Prinzipiell könnten unterschiedliche Nebenwirkungsprofile Behandlungen für solche Patienten ermöglichen, für die andere Arzneimittel mit demselben Wirkprinzip unverträglich sind. Dies wird aber bereits standardmäßig ohnehin durch das IQWiG als zusätzlicher Nutzen berücksichtigt.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Geheilt statt behandelt»
Представляем Вашему вниманию похожие книги на «Geheilt statt behandelt» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Geheilt statt behandelt» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.