Heinz Wiendl - Multiple Sklerose
Здесь есть возможность читать онлайн «Heinz Wiendl - Multiple Sklerose» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на немецком языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Multiple Sklerose
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Multiple Sklerose: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Multiple Sklerose»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Multiple Sklerose — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Multiple Sklerose», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Merke
Zerebrale Läsionen schließen das Vorliegen einer NMO sogar dann nicht aus, wenn sie die Barkhof/McDonald-Kriterien für eine MS erfüllen.
Liquor
In der Liquoranalyse kann bei der NMO während eines akuten Schubereignisses eine lymphozytäre und neutrophile Pleozytose zwischen 50/µl und 1.000/µl beobachtet werden, während die Zellzahl in der Remission oftmals unauffällig oder nur leichtgradig erhöht ist (Wingerchuk et al. 1999). Schwere Pleozytosen mit > 100 Zellen/µl sind dennoch eher selten (Jarius et al. 2011). Oligoklonale Banden im Liquor, die bei ca. 95 % der MS-Patienten zu finden sind (Ebers und Paty 1980; McLean et al. 1990), kommen nur in ca. 15–30 % der NMO-Fälle vor (Wingerchuk et al. 1999; de Sèze 2002; O’Riordan et al. 1996; Bergamaschi 2004; Jarius et al. 2011) und verschwinden häufig im Krankheitsverlauf (35–40 %) (Wingerchuk et al. 1999; Ghezzi 2004; Bergamaschi 2004). Eine positive intrathekale MRZ-Reaktion (Masern, Röteln, Varizella Zoster) im Rahmen einer unspezifischen B-Zell-Aktivierung, wie bei den meisten MS-Patienten zu sehen, ist bei der NMO nicht nachweisbar (Jarius et al. 2008; Reiber 1998; Meinl et al. 2006). Eine japanische Studie zeigte des Weiteren bei Patienten mit einer möglichen NMO einen erhöhten Protein 14-3-3-Liquorspiegel, der als Korrelat des ausgeprägten neuronalen Untergangs im Schub zu sehen ist (Satoh 2003).
Serologische Biomarker- NMO-IgG/Aquaporin-4 AK
Serologische Biomarker tragen zur frühen Diagnostik der NMO und Unterscheidung der NMO von der MS bei. 2004 wurde erstmals ein Antikörper nachgewiesen, der an den Astrozytenendfüßen bindet (NMO-IgG) (Lennon et al. 2004). Im Verlauf wurde gesichert, dass es sich bei dem Ziel-Antigen um Aquaporin-4 handelt, den häufigsten Wasserkanal im ZNS (Lennon et al. 2005). Der Nachweis führte zu einer Verbesserung der Diagnosestellung und Revision der Diagnosekriterien 2006, die 2015 erneut überarbeitet wurden (Wingerchuk et al. 2015,  Abb. 1.2)
Abb. 1.2)
Aquaporin-4 ist ein bidirektionaler Wasserkanal, der zur Unterfamilie der mamillären Aquaporine gehört, welche impermeabel für Anionen und Glycerol sind. Es befindet sich
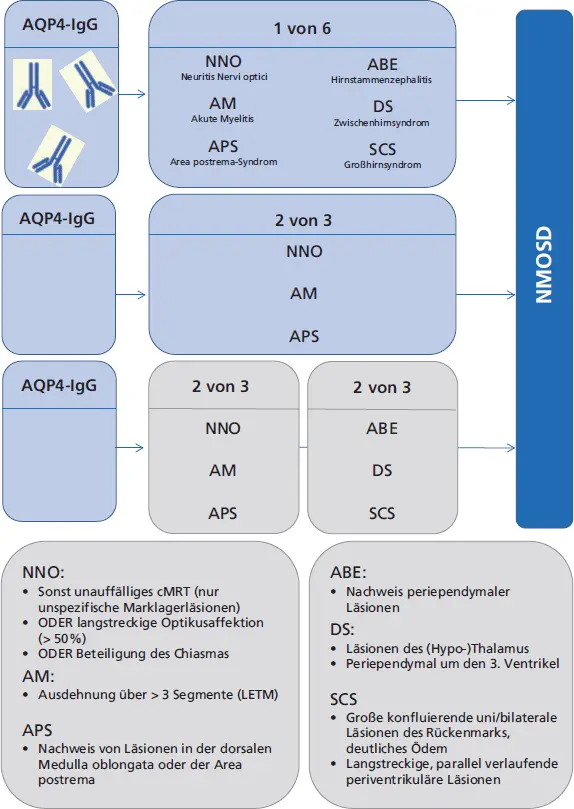
Abb. 1.2: Schematische Darstellung der Diagnosekriterien (Pfeuffer et al. 2017, S. 16).
in den perivaskulären astrozytären Endfüßchen, sowie in der Pia mater, dem Plexus choroideus und dem Ependym, ist aber auch in der Niere (distale Sammelröhrchen) und im Magen (Parietalzellen) (Guo et al. 2017). In Neuronen, Oligodendrozyten oder choroidalen Epithelzellen ist es nicht nachweisbar (Jung 1994). Studien konnten belegen, dass Aquaporin-4 im Sehnerv, der grauen Substanz des Rückenmarks und auch im Hirnstammbereich stärker exprimiert wird, was mit den typischen Läsionsregionen korreliert (Roemer et al. 2007; Pittock und Lucchinetti 2016). Tierexperimentell scheint ein Fehlen des zerebralen, perivaskulären Aquaporin-4 zu einem leichten Anschwellen der astrozytären Endfüßchen zu führen, allerdings ohne nachweisbares funktionelles Defizit. Die entsprechenden defizienten Mäuse neigen zu einer geringeren Ödembildung nach hyposmotischem Stress und nach experimenteller Ischämie (Manley 2000). In anderen Tierexperimenten entwickelten Alpha-syntrophin-Null Mäuse, die nahezu Aquaporin-4 depletiert sind, bei der artifiziellen Anfallsinduktion schwerere Anfälle, vermutlich aufgrund eines gestörten Kaliummetabolismus, da Aquaporin-4 räumlich eng mit einem koexprimierenden Kaliumkanal (Kir4.1) assoziiert ist (Amiry-Moghaddam et al. 2003).
Die Beteiligung von Aquaporin am Pathomechanismus der Erkrankung konnte in vitro belegt werden. Hier führte die Applikation von aufgereinigten Aquaporin-4-AK sowohl zum komplement-abhängigen als auch zum komplement-unabhängigen Astrozytenuntergang (Nishiyama et al 2016).
Neben der initial angewandten gewebeabhängigen Nachweismethode stehen inzwischen sowohl zellbasierte (ICC, FACS, cell-ELISA) als auch proteinbasierte Assays zum Nachweis der Antikörper zur Verfügung (RIPA/FIPA, WB, ELISA) (Jarius und Wildemann 2013). Zur Diagnostik sind zellbasierte Verfahren mit Bestätigungstest ratsam, da diese in gepoolten Analysen die höchste Sensitivität von 76,7 % aufwiesen (Wingerchuk et al. 2015; Jarius und Wildemann 2013; Waters et al. 2012; Sato et al. 2013), die Spezifität erreicht dagegen bei 85–100 % (Wingerchuk et al. 2006; Lennon et al. 2004; Jarius und Wildemann 2007; Nakashima et al. 2006; Jarius und Wildemann 2013).
Bei Aquaporin-4-Antikörper-positiven Kindern wurden neben den typischen NMO-Symptomen in 45 % der Fälle zerebrale Symptome wie eine Enzephalopathie oder Krampfanfälle festgestellt (McKeon et al. 2008), hier wurde eine eigene Krankheitsentität postuliert.
Merke
Bei einem großen Teil der betroffenen Patienten können NMO-IgG (Aquaporin-4-Antikörper) nachgewiesen werden. Ist der Nachweis jedoch unmöglich, schließt das eine NMO nicht aus.
Der Nachweis von Aquaporin-4-AK im Liquor wird aktuell noch kritisch diskutiert, denn möglicherweise handelt es sich um extrathekal produzierte Antikörper. Der Test wird daher nicht in der klinischen Routine angewandt (Jarius und Wildemann 2013). Ein Zusammenhang der Titerhöhe mit dem klinischen Verlauf konnte nicht nachgewiesen werden (Kessler et al. 2017).
Beim Monitoring in der klinischen Praxis werden NMO-IgG bisher nicht als Parameter bestimmt, um die Therapieeffizienz zu evaluieren. Retrospektive Analysen zeigen allerdings, dass Antikörper bei initial seropositiven Patienten unter einer immunsuppressiven Therapie im schubfreien Intervall nicht nachweisbar sind (Weinstock-Guttman et al. 2006), sodass eine dauerhaft NMO-IgG-Negativität möglicherweise ein Korrelat für einen guten therapeutischen Effekt darstellen kann. Ebenso konnte gezeigt werden, dass der Titer unter einer erfolgreichen Rituximabtherapie sank, während dies vor Schüben und bei Non-Respondern nicht geschah (Valentino et al. 2016; Jarius et al. 2008).
Histologie und Pathogenese
Histopathologisch lassen sich zwei unterschiedliche Muster nachweisen. In der akuten Phase findet sich in den klassischen Läsionen eine ausgeprägte konfluierende oder perivaskuläre, entzündliche Demyelinisierung mit hochgradigem axonalen Schaden. Man findet die Veränderungen sowohl in der weißen als auch der grauen Substanz. Nekrosen, Hohlraumbildung und ein Verlust an Oligodendrozyten sind zu beobachten. In dem zweiten Läsionstyp finden sich Vakuolen im Myelin sowie eine reaktive Astrozyten- und Mikrogliaaktivierung, eine granulozytäre Entzündung mit nur geringem axonalen Schaden (Pittock und Lucchinetti 2016). Ob dieser zweite Läsionstyp ebenfalls in Nekrosen mündet oder reversibel sein kann, ist ungeklärt. (Mandler et al. 1993; Marignier et al. 2010; Roemer et al. 2007). Die entzündlichen Infiltrate setzten sich hauptsächlich aus einer Ansammlung von Makrophagen, T- und B-Lymphozyten, Plasmazellen sowie neutrophilen und eosinophilen Granulozyten zusammen. Auch lassen sich perivaskuläre Immunglobuline (vor allem IgM) und Komplementfaktoren nachweisen ebenso wie fibrotisch verdickte und hyalinisierte Gefäße (Lucchinetti et al. 2002; Pittock und Lucchinetti 2016). Insbesondere die vaskulozentrische Ablagerung von Komplement und Immunglobulinen, die vaskuläre Fibrose und die Beteiligung von Eosinophilen unterscheidet die Läsionen von MS-Läsionen. Ein weiterer Unterschied zur MS zeigt sich anhand der Astrozyten . Während die MS unter anderem durch eine ausgeprägte Astrozytose gekennzeichnet ist (Holley 2003; Ayers 2004), kommt es bei der NMO zu einer teilweise verminderten Expression des Astrozytenmarkers GFAP als auch von Aquaporin 1 und 4 (Roemer et al. 2007; Lucchinetti et al. 2014). Histopathologisch kommt es zu einem raschen Astrozytenuntergang, gefolgt von einer frühen Schädigung von Oligodendrozyten und einem axonalen Schaden (Pittock und Lennon 2008).
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Multiple Sklerose»
Представляем Вашему вниманию похожие книги на «Multiple Sklerose» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Multiple Sklerose» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.