Heinz Wiendl - Multiple Sklerose
Здесь есть возможность читать онлайн «Heinz Wiendl - Multiple Sklerose» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на немецком языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Multiple Sklerose
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Multiple Sklerose: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Multiple Sklerose»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Multiple Sklerose — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Multiple Sklerose», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
2. Antiinflammatorische T H2-Lymphozyten, deren Zytokinmuster durch antiinflammatorische Zytokine, wie beispielsweise IL-4 oder IL-10, charakterisiert ist und einen hemmenden Einfluss auf die Immunantwort ausübt.
3. T H17-Zellen, charakterisiert durch die Freisetzung von IL-17, eine Subpopulation von T-Zellen, denen eine besondere Bedeutung bei der Krankheitsinitiierung eingeräumt wird.
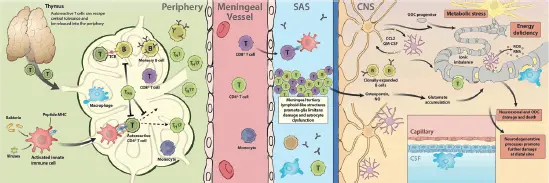
Abb. 3.4: Aktivierung und Rolle der T-Lymphozyten bei MS und EAE (© Heike Blum).
3.4.2 Die Rolle von B-Lymphozyten
Der Nachweis oligoklonaler Banden im Liquor von MS-Patienten ist ein typischer Befund, der auf eine B-Zell-/Plasmazellaktivierung hinweist. Dennoch wurden B-Zellen im Kontext der Immunpathogenese der MS lange Zeit eher vernachlässigt. Die entzündlichen Infiltrate einer aktiven MS-Läsion enthalten wenige B-Lymphozyten oder Plasmazellen im Verhältnis zu T-Zellen. Bei Patienten mit sekundär chronisch progredienter MS konnten jedoch B-Zell-Follikel in den Meningen gefunden werden (Serafini et al. 2004). Der Nachweis von B-Zellen in der MS-Läsion lässt natürlich spekulieren, ob diese Zellen an der Entzündung durch die Freisetzung myelinspezifischer Autoantikörper partizipieren, allerdings sind bis jetzt in der MS im Gegensatz zu NMOSD keine Antikörper gefunden worden, die diese Theorie stützen. Für deren Existenz spricht jedoch die Wirksamkeit der Plasmaseparation.
Eine weitere möglicherweise zur MS beitragende B-Zell-Funktion neben der Antigenpräsentation und der Antikörperproduktion ist die Produktion von proinflammatorischen Zytokinen (Bittner et al. 2017). Zusätzlich spielen regulatorische B-Zellen (Bregs) offenbar eine wichtige Rolle bei der Sicherheit und Wirksamkeit von B-Zell-Therapien. Nach Rituximab-Infusion finden sich Bregs mit einer erhöhten CD38- und CD5-Expression, die nach Aktivierung weniger proinflammatorische Zytokine produzieren. Aktuell ist als B-Zell-depletierende Therapie Ocrelizumab zugelassen, Rituximab wird seit langem mit Erfolg off-label eingesetzt, mit Ofatumumab könnte möglicherweise ein weiterer B-Zell-depletierender Antikörper zugelassen werden.
3.4.3 Terminierung der Immunreaktion
Das Immunsystem verfügt über zahlreiche Mechanismen, um die massive Expansion zellulärer und löslicher Mediatoren zu kontrollieren. Sobald das Ziel-Antigen eliminiert wurde, werden die Effektorzellen nicht länger benötigt und es vollzieht sich ein programmierter Zelltod, Apoptose genannt (Gold et al. 1997; Zipp et al. 1999).
Das Überleben von Lymphozyten hängt von der Balance den Zelltod fördernder und hemmender Faktoren ab. Die Apoptose wird von einigen Mitgliedern der intrazellulären Bcl-2-Familie verhindert (Hengartner 2001), wohingegen andere Mechanismen Apoptose induzieren, beispielsweise durch die Interaktion des Zelloberflächenrezeptors Fas mit dem Liganden Fas-Ligand, der zur TNF-Familie gerechnet wird (Krammer 2000). Glucocortikoide können die Apoptose von T-Zellen induzieren, eine Beobachtung mit therapeutischer Implikation für die MS (Gold et al. 2001).
3.4.4 Das andere Gesicht der Entzündung
Die bisherige Ansicht, dass die lokale Entzündungsreaktion im ZNS nur deletäre Elemente in sich birgt, hat sich in den vergangenen Jahren verändert. Neuere Untersuchungen legen nahe, dass Entzündungszellen nicht nur hilfreich für das Gewebe sind, sondern möglicherweise sogar neuroprotektive Effekte mediieren können. So konnte gezeigt werden, dass der sog. brain-derived neurotrophic factor (BDNF), ein neuroprotektives Molekül, in MS-Läsionen von invadierenden T-Lymphozyten exprimiert wird (Kerschensteiner et al. 1999; Stadelmann et al. 2002) (Hohlfeld et al. 2001; Kerschensteiner et al. 2003).
Zusätzlich wird seit einigen Jahren die Bedeutung regulatorischer B- und T-Zellen (Bregs und Tregs) untersucht. Tregs sind wichtig, um die Selbsttoleranz zu erhalten, Autoimmunität zu verhindern und die Immunreaktion auf Stimuli zu begrenzen. Sie sind immunologisch durch die Expression von CD25 und FAxP3 definiert (Fontenot et al. 2003).
Im Tiermodell konnte gezeigt werden, dass bei der EAE in Phasen der klinischen Stabilität die Zahl der Tregs in den Läsionen zunimmt. Treg-Eliminierung führt folgerichtig zu einer Krankheitsverschlechterung (Duffy et al. 2018). Bei MS-Patienten wurde festgestellt, dass Tregs phänotypisch verändert und funktionell eingeschränkt sind (Haas et al. 2007; Viglietta et al. 2004). Wie Untersuchungen erwiesen, können neben Tregs auch aktivierte B-Zellen Immunreaktionen durch Zytokinproduktion unterdrücken, insbesondere mittels IL-10, aber auch IL-35 oder TGF-β (Lampropoulou et al. 2010; Rosser et al. 2015). Damit ist die Einwanderung von Lymphozyten nicht notwendigerweise ein destruktives Ereignis für das ZNS, sondern kann auch protektive Eigenschaften haben (Zozulya und Wiendl 2008).
3.4.5 Axonale Schädigung und Neurodegeneration
Das Ausmaß an axonalem Verlust in der Pathogenese der MS ist größer als zuvor angenommen und trägt offenbar maßgeblich zur klinischen Progression der Erkrankung bei (Medana und Esiri 2003; Fisniku et al. 2008). Interessanterweise zeigt sich, dass das Ausmaß axonaler Schädigung besonders in den ersten Jahren einer klinisch floriden MS hoch ist (Kuhlmann et al. 2002; Schirmer et al. 2013; Pfeifenbring et al. 2015). Die zugrunde liegenden molekularen Mechanismen sind derzeit noch nicht vollständig geklärt, doch es scheint ein multifaktorieller Prozess zu sein (Neumann 2003). Neben der Entzündung gibt es Hinweise darauf, dass sowohl die Destruktion von Kalium- und Kalziumkanälen, oder des axonalen Transports, eine mitochondriale Fehlfunktion (Dutta et al. 2006; Campbell et al. 2011; Campbell at al. 2014) als auch oxidativer Stress (Fischer et al. 2012; Haider et al. 2011) eine Rolle spielen.
Eine wesentliche Frage, die es zu beantworten gilt, lautet, ob und wie sich axonale Schädigung und Demyelinisierung bedingen (Irvine und Blakemore 2008). Eine mögliche Erklärung wäre, dass die Myelinhülle Axone vor direkten Attacken des Immunsystems zu schützen vermag und somit die primär stattfindende Demyelinisierung eine sekundäre Schädigung von Axonen bedingt (Rodriguez 2003). Dafür spricht ein gewisser protektiver Effekt durch eine Remyelinisierung (Schultz et al. 2017). Eine alternative Hypothese geht davon aus, dass eine Fehlfunktion der Oligodendrozyten ausreicht, um eine axonale Degeneration zu induzieren (Lappe-Siefke et al. 2003).
3.4.6 Infektiöse Erreger und Multiple Sklerose
Der Zusammenhang zwischen infektiösen Erregern (insbesondere Viren und Bakterien) und der MS ist seit jeher Gegenstand intensiver Diskussion. Eine Reihe indirekter Hinweise suggeriert bis heute den Einfluss eines solchen »Umweltfaktors«: das Vorliegen von Hochrisikogebieten, dokumentierte MS-Epidemien, die Isolierung bestimmter Viren bei MS-Patienten (z. B. Herpes-simplex-Virus, HTLV-1, humaner Herpesvirus Typ 6, Multiple-Sklerose-assoziiertes Retrovirus, MSRV, Epstein-Barr-Virus, EBV), serologische Studien mit dem Nachweis Virus-spezifischer Antikörper oder T-Zellen, der Nachweis viraler Genome im ZNS-Gewebe sowie Virus-Tiermodelle der MS. Allerdings hat sich bis heute keiner der publizierten Erreger als singuläres infektiöses »MS-Agens« identifizieren lassen, eine Tatsache, die sich in der Widersprüchlichkeit des Schrifttums zu Virusisolierungen oder Nachweisen von viralen Genomen im MS-Gewebe widerspiegelt (Übersichtsarbeiten bei Cermelli et al. 2000; Meinl 1999; Gilden 2005). Die überzeugendsten Daten existieren bislang für den Zusammenhang von EBV mit der MS. Nahezu 100 % der MS-Patienten sind seropositiv für EBV, zudem zeigen sich in epidemiologischen Studien klare Hinweise für einen Zusammenhang einer EBV-Infektion mit dem Beginn einer MS (DeLorenze et al. 2006; Levin et al. 2010). Wie sich die EBV-Infektion im Pathomechanismus auswirkt, ist allerdings weiterhin unklar. So werden sowohl molekulare Mimikry als auch eine EBV-induzierte B-Zelltransformation diskutiert (Lang et al. 2002; Tracy et al. 2012). Bewertet man die bekannten epidemiologischen und genetischen Erkenntnisse, so haben virale Agentien oder andere Umweltfaktoren zusammen mit den teilweise bekannten genetischen Komponenten offenbar Einfluss auf die Erkrankungssuszeptibilität bzw. den Erkrankungsausbruch. Dieser Schluss wird durch Daten unterstützt, die zeigen, dass bis zu 25 % der akuten Schübe durch akute virale Infektionen ausgelöst werden (Sibley et al. 1985) oder Virusinfektionen dem Erkrankungsbeginn vorangehen können (z. B. Ascherio et al. 2001). Es bleibt derzeit ungeklärt, ob 1. Viren eine notwendige Voraussetzung für die Initiierung oder Reaktivierung einer MS sind oder ob 2. primär eine Autoimmunreaktion gegen ZNS-Gewebe vorliegt, die potenziell durch virale (Ko-)Infekte moduliert wird.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Multiple Sklerose»
Представляем Вашему вниманию похожие книги на «Multiple Sklerose» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Multiple Sklerose» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.