Stephen Winters-Hilt - Informatics and Machine Learning
Здесь есть возможность читать онлайн «Stephen Winters-Hilt - Informatics and Machine Learning» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Informatics and Machine Learning
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Informatics and Machine Learning: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Informatics and Machine Learning»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Discover a thorough exploration of how to use computational, algorithmic, statistical, and informatics methods to analyze digital data Informatics and Machine Learning: From Martingales to Metaheuristics
ad hoc, ab initio
Informatics and Machine Learning: From Martingales to Metaheuristics
Informatics and Machine Learning — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Informatics and Machine Learning», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
The next program, cleverly named prog2.py, will build off the code devised previously, with the file i/o operation now “lifted” into a subroutine for safer encapsulation (to avoid scope errors, etc.) and to avoid the confusing clutter of copying and pasting such a large block of code repeatedly that would be required otherwise. By now, this has hopefully made a convincing case for why subroutines are a big deal in the evolution of software engineering constructs (and the computer languages that implement them). Further discussion is given in the comments in the code.
------------------------ prog2.py --------------------------- #!/usr/bin/python import numpy as np import math import re # from prior code we carry over the subroutines: # shannon, count_to_freq, Shannon_order; # with prototypes: # def shannon( probs ) with usage: # value = shannon(probs) # print(value) # # def count_to_freq( counts ) with usage # probs = count_to_freq(rolls) # print(probs) # # def shannon_order( seq, order ) with usage: # order = 8 # maxcounts = 4**order # print "max counts at order", order, "is =", maxcounts # val = math.log(maxcounts) # shannon_order(sequence,order) # shannon_order prints entropy # print "The max entropy would be log(4**order) = ", val # New code is now created to have subroutines for text handling. # There are two types of text-read, one for genome data in "fasta" # format (gen_fasta_read) and one for generic format (gen_txt_read): def gen_txt_read( text ): if (text == ""): text = "Norwalk_Virus.txt" fo = open(text, "r+") str = fo.read() fo.close() pattern = '[acgt]' result = re.findall(pattern, str) # seqlen = len(result) return result #usage null="" gen_array = gen_txt_read(null) # defaults, uses Norwalk_Virus.txt genome sequence = "" sequence = sequence.join(gen_array) Norwalk_sequence = sequence; seqlen = len(gen_array) print "The sequence length of the Norwalk genome is:", seqlen def gen_fasta_read( text ): if (text == ""): text = "EC_Chr1.fasta.txt" slurp = open(text, 'r') lines = slurp.readlines() sequence = "" for line in lines: pattern = '>' test = re.findall(pattern,line) testlen = len(test) if testlen>0: print "fasta comment:", line.strip() else: sequence = sequence + line.strip() slurp.close() pattern = '[acgtACGT]' result = re.findall(pattern, sequence) return result #usage null="" fasta_array = gen_fasta_read(null) # defalus, uses e.coli genome sequence = "" sequence = sequence.join(fasta_array) EC_sequence = sequence seqlen = len(fasta_array) print "The sequence length of the e. coli Chr1 genome is:", seqlen print "Doing order 8 shannon analysis on e-coli:" order = 6 shannon_order(EC_sequence,order) --------------------- prog2.py end --------------------------
As mentioned previously, when comparing two probability distributions on the same set of outcomes, it is natural to ask if they can be compared in terms of the difference in their scalar‐valued Shannon entropies. Similarly, there is the standard manner of comparing multicomponent features by treating them as points in a manifold and performing the usual Euclidean distance calculation generalized to whatever dimensionality of the feature data. Both of these approaches are wrong, especially the latter, when comparing discrete probability distributions (of the same dimensionality). The reason being, when comparing two discrete probability distributions there are the additional constraint on the probabilities (sum to 1, etc.), and the provably optimal difference measure under these circumstances, as described previously, is relative entropy. This will be explored in Exercise 3.5, so some related subroutines are included in the first addendum to prog2.py:
--------------------- prog2.py addendum 1-------------------- # We can use the Shannon_order subroutine to return a probability array # for a given sequence. Here are the probability arrays on 3-mers # (ordered alphabetically): Prob_Norwalk_3mer = shannon_order(Norwalk_sequence,3) Prob_EC_3mer = shannon_order(EC_sequence,3) # the standard Euclidean distance and relative entropy are given next def eucl_dist_sq ( P , Q ): Pnum = len(P) Qnum = len(Q) if Pnum != Qnum: print "error: Pnum != Qnum" return -1 euclidean_distance:squared = 0 for index in range(0, Pnum): euclidean_distance:squared += (P[index]-Q[index])**2 return euclidean_distance:squared # usage value = eucl_dist_sq(Prob_Norwalk_3mer,Prob_EC_3mer) print "The euclidean distance squared between EC and Nor 3mer probs is", value # P and Q are probability arrays, meaning components are positive definite # and sum to one. If P and Q are proability arrays, can compare them in # terms of relative entropy (not Euclidean distance): def relative_entropy ( P , Q ): Pnum = len(P) Qnum = len(Q) if Pnum != Qnum: print "error: Pnum != Qnum" return -1 rel_entropy = 0 for index in range(0, Pnum): rel_entropy += P[index]*math.log(P[index]/Q[index]) return rel_entropy # usage value1 = relative_entropy(Prob_Norwalk_3mer,Prob_EC_3mer) print "The relative entropy between Nor and EC 3mer probs is", value1 value2 = relative_entropy(Prob_EC_3mer,Prob_Norwalk_3mer) print "The relative entropy between EC and Nor 3mer probs is", value2 sym = (value1+value2)/2 print "The symmetrized relative entropy between EC and Nor 3mer probs is", sym -------------------- prog2.py addendum 1end ------------------
Recall that the definition of mutual information between two random variables, { X , Y } is simply the relative entropy between P ( X , Y ) “ P ” and P ( X ) P ( Y ) “ p ”, and this is implemented in addendum #2 to prog2.py:
------------------- prog2.py addendum 2 ---------------------- # the mutual information between P(X,Y) and p(X)p(Y) requires passing two # prob arrays: P and p, where |P| = |p|^2 is relation between # of terms: def mutual_info ( P , p ): Pnum = len(P) pnum = len(p) if Pnum != pnum*pnum: print "error: Pnum != pnum*pnum" return -1 mi = 0 for index in range(0, Pnum): row = index/pnum column = index%pnum mi += P[index]*math.log(P[index]/(p[row]*p[column])) return mi #usage Prob_EC_2mer = shannon_order(EC_sequence,2) Prob_EC_1mer = shannon_order(EC_sequence,1) mutual_info(Prob_EC_2mer,Prob_EC_1mer) ----------------- prog2.py addendum 2 end -------------------
3.2 Codon Discovery from Mutual Information Anomaly
As mentioned previously, mutual information allows statistical linkages to be discovered that are not otherwise apparent. Consider the mutual information between nucleotides in genomic data when different gap sizes are considered between the nucleotides as shown in Figure 3.1a. When the MI for different gap sizes is evaluated (see Figure 3.1b), a highly anomalous long‐range statistical linkage is seen, consistent with a three‐element encoding scheme (the codon structure is thereby revealed) [1, 3].
To do the computation for Figure 3.1, the next subroutine (prog2.py addendum 3) is a recycled version of the code for the dinucleotide counter described previously (prog1.py addendum 6), except that now the two bases in the sample window have a fixed gap size between them of the indicated size. Before, with no gap, the gapsize was zero.
------------------- prog2.py addendum 3 --------------------- def gap_dinucleotide( seq, gap ): stats = {} pattern = '[acgtACGT]' result = re.findall(pattern, seq) seqlen = len(seq) probs = np.empty((0)) if (1+gap>seqlen): print "error, gap > seqlen" return probs for index in range(1+gap,seqlen): xmer = result[index-1-gap]+result[index] if xmer in stats: stats[xmer]+=1 else: stats[xmer]=1 counts = np.empty((0)) for i in sorted(stats): counts = np.append(counts,stats[i]+0.0) probs = count_to_freq(counts) return probs # usage: for i in range(0,25): gap_probs = gap_dinucleotide(EC_sequence,i) val=mutual_info(gap_probs,Prob_EC_1mer) print "Gap", i, "has mi=", val ----------------- prog2.py addendum 3 end -------------------- 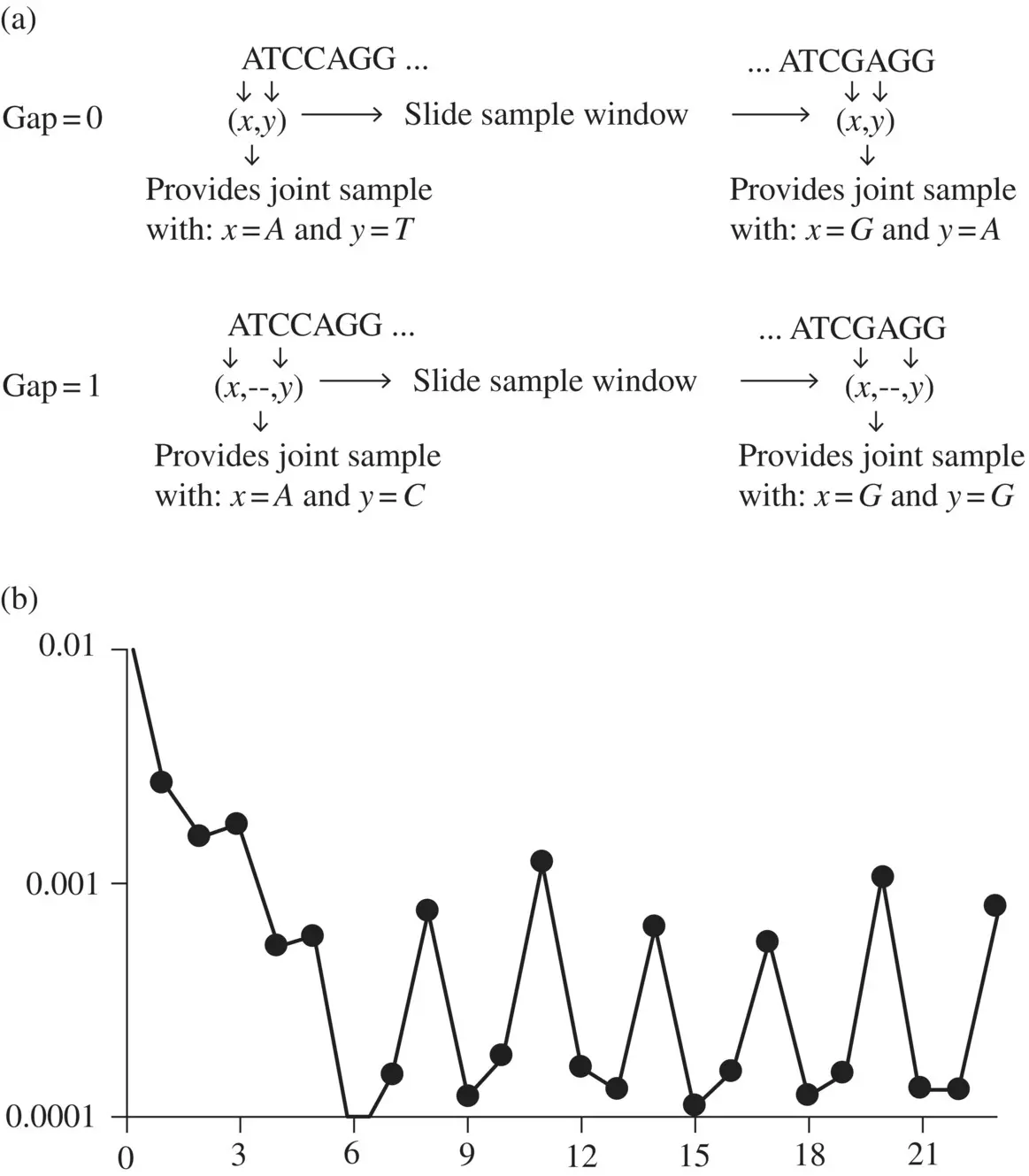
Интервал:
Закладка:
Похожие книги на «Informatics and Machine Learning»
Представляем Вашему вниманию похожие книги на «Informatics and Machine Learning» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Informatics and Machine Learning» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.