Polar Organometallic Reagents
Здесь есть возможность читать онлайн «Polar Organometallic Reagents» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Polar Organometallic Reagents
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Polar Organometallic Reagents: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Polar Organometallic Reagents»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Polar Organometallic Reagents
Polar Organometallic Reagents
Polar Organometallic Reagents — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Polar Organometallic Reagents», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Prior to the advent of ate chemistry, attempts to overcome the problem of organometallic nucleophilicity focused on the deployment of other metals. For example, in 1989, Eaton et al. reported the selective magnesiation of alkyl benzoates using sterically demanding magnesium amide 12( Scheme 1.5) [29], suggesting the possibility of highly chemoselective conversion to e.g. 13in the presence of ester and amide moieties. This protocol was used to ortho ‐carboxylate methyl benzoate, giving 14in 81% yield.
Using a similar thesis, 1‐substituted indole derivatives have been deprotonated using a magnesium diamide to give magnesioindoles, which were then successfully reacted with electrophiles. The compatibility of the magnesiated intermediates with a range of electrophilic functional groups was examined. For example, methyl 1‐phenylsulfonylindole‐3‐carboxylate was treated with ( i ‐Pr 2N) 2Mg 15followed by iodine or benzaldehyde to give 2‐iodo derivative 16and alcohol 17in the respective yields 85 and 93% ( Scheme 1.6) [30]. 1‐Phenylsulfonylindole‐3‐carbonitrile was also tested in this iodination using i ‐Pr 2NMgBr 18at the outset. In a similar vein, ethyl n ‐thiophenecarboxylate ( n = 2, 3) has been selectively deprotonated with retention of the ester group, using i ‐Pr 2NMgCl 19to give 2,5‐ and 2,3‐disubstituted thiophenes, respectively [31]. Meanwhile, the selective deprotonation of pyridine carboxamides and carbamates in conjunction with the more sterically congested magnesium amide TMPMgCl 20has been reported [32]. Deprotonative magnesiation has been further investigated by Knochel and its scope and limitations have been the subject of review [33].
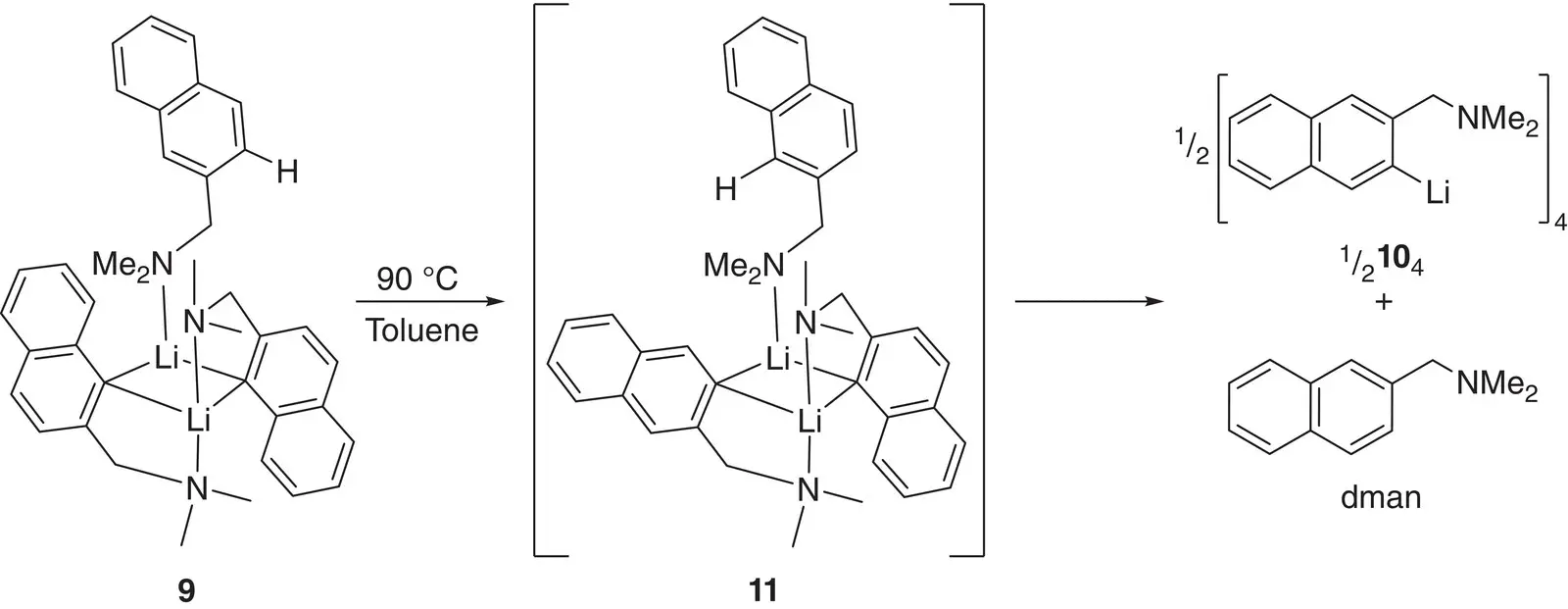
Scheme 1.4 The rearrangement of [2‐(Me 2NCH 2)C 10H 6Li‐1] 2(dman) 9in hot toluene.
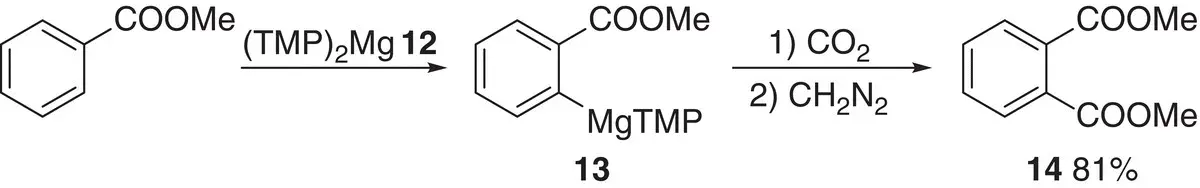
Scheme 1.5 Selective magnesiation of an alkyl benzoate using magnesium amide 12.
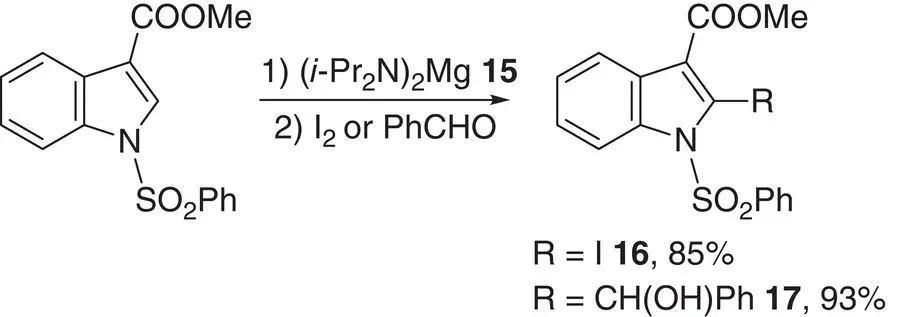
Scheme 1.6 Treatment of methyl 1‐phenylsulfonylindole‐3‐carboxylate with ( i ‐Pr 2N) 2Mg 15en route to 2‐iodinated 16and alcohol 17.
Detailed elucidation of the products of directed aromatic magnesiation has been enabled using air‐sensitive crystallographic techniques. The same is true of precursors to deprotonation, with for example, the constitution of the deprotonating agents Grignard and Hauser bases probed. In these contexts, upon exposure to THF, MeMgCl 21was found to form MeMg 2(μ‐Cl) 3(THF) 4‐6 22, but to dimerize to give Me 2Mg 4Cl 6(THF) 6 23in the solid‐state [34]. In contrast, externally solvated alkyl‐and halo(amido)magnesiums formed straightforward monomers and dimers [35]. Moving to ortho ‐magnesiation, the bisamide 12has been used to smoothly react boron‐substituted benzenes. This work evolved from the advent of TMP‐bases as ortho ‐metalating agents and enabled the ortho ‐magnesiation of borylbenzenes via internal reaction of an N ‐magnesiated intermediate 24. The resulting C , N ‐magnesiate 25was then electrophilically quenched using Me 2SO 4to provide methylated product 26that could be converted to a pinacolate ( Scheme 1.7). The structure of a representative ortho C , N ‐magnesiated intermediate proved to be a TMP‐intercepted spirocycle (in effect, a dimer of 25( 12); Figure 1.3) [36].
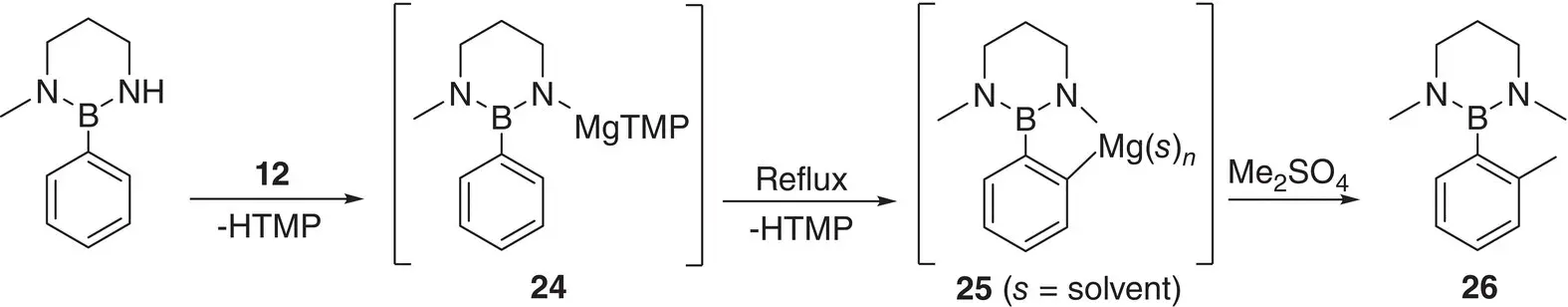
Scheme 1.7 Ortho ‐magnesiation of a borylbenzene via internal reaction of N ‐magnesiated intermediate 24to give C , N ‐magnesiate 25.
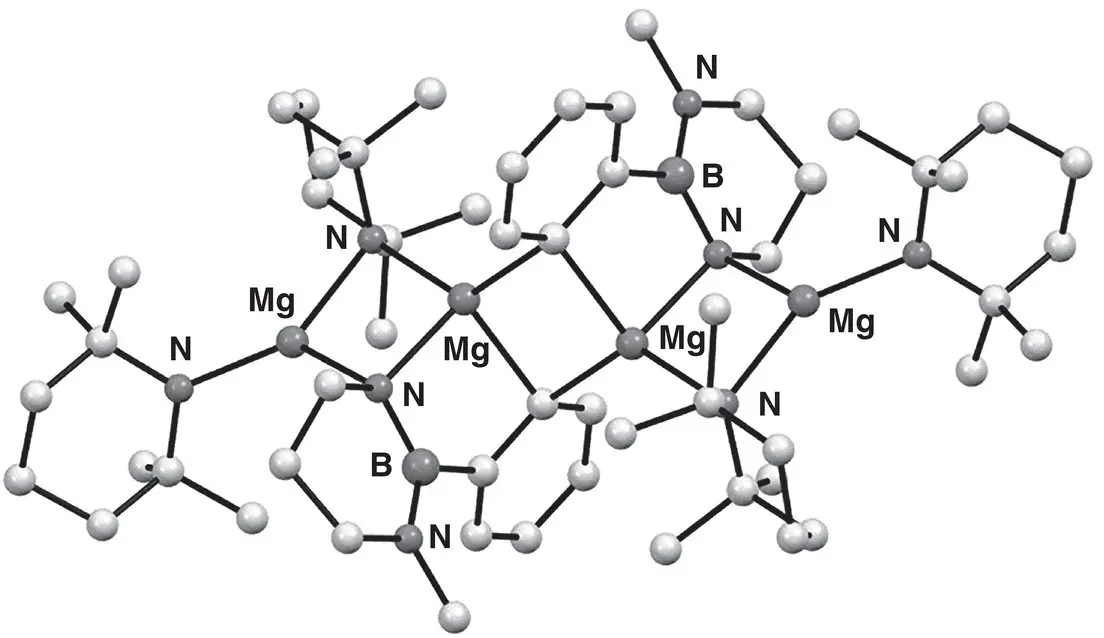
Figure 1.3 Structure of the ortho C , N ‐magnesiated dimer of 25( 12).
Source : Adapted from Kawachi et al. [36].
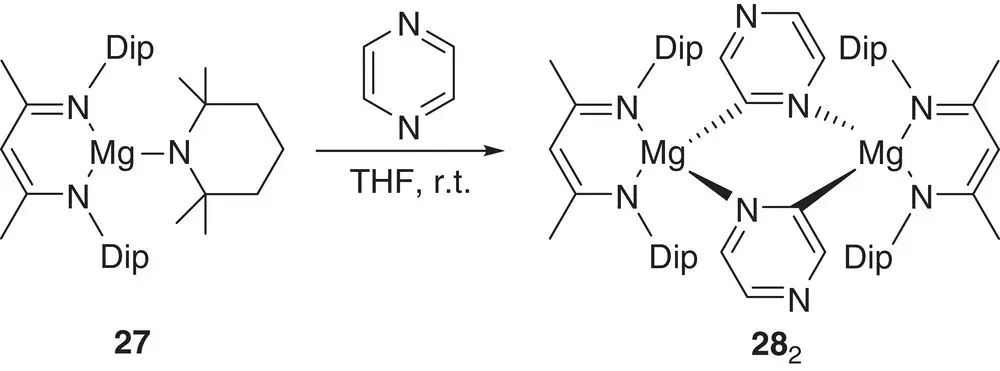
Scheme 1.8 The use of ( DipNacnac)MgTMP 27in pyrazine deprotometalation.
β‐Diketiminate magnesium complexes have been developed very recently and applied to the regioselective magnesiation of aromatics. Initially, ( DipNacnac)MgTMP 27( DipNacnac = DipNC(Me)CHC(Me)NDip; Dip = 2,6‐( i‐ Pr) 2‐C 6H 3), which combines kinetic base TMP with a sterically demanding spectator β‐diketiminate, was reacted with pyrazine at room temperature to quantitatively yield ( DipNacnac)Mg(C 4H 3N 2) 28( Scheme 1.8) [37]. This work was then extended to using the kinetic TMP base to trap sensitive fluoroaryl anions for deployment in Negishi cross‐coupling. This trapping behavior contrasted with that of the kinetic‐amide‐lacking organyl analogue ( DipNacnac)Mg(R)(THF) 29(R = n ‐Bu, Ph, benzofuryl), which was shown to be effective in the chemically interesting [38, 39] magnesiation of perfluorinated aromatics by C–F bond alkylation/arylation [40].
1.2.2 Bimetallic Bases
1.2.2.1 Group 1/1 Reagents
An alternative approach to directed aromatic metalation has focused not upon replacing the alkyllithium base but on activating it. Two methods by which to achieve this were developed some time ago. The first was centered on TMEDA‐activation (TMEDA = N , N , N′ , N′ ‐tetramethylethylenediamine) and the second involved the use of tert ‐butoxide‐complexed alkyllithium reagents in the form of LICKOR superbases. The former route was employed to achieve site‐selective deprotonation with different selectivity to that achieved using the alkyllithium alone. Meanwhile, whereas unimetallic superbases are known [41], 1966–1967 saw the introduction by Lochmann [42] and Schlosser [43] of heterobimetallic (Li–Na/K) superbases. Subsequently extended to incorporate a range of alkyllithium adducts of potassium alkoxides [44], the most widely known example deploys traditional organolithium reagents in tandem with KO t ‐Bu. Such heterobimetallic systems have shown enormous reactivity toward deprotonative metalation [45, 46]. As such, they enable the smooth deprotometalation of low acidity hydrocarbons [47] and weakly activated or nonactivated benzene derivatives [48] with, in some cases, unique regioselectivity [49] and also the facility for multideprotonation [50]. Whilst the synthetic importance of heterobimetallic superbases was quickly established, the characterization of such air‐sensitive materials lagged behind.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Polar Organometallic Reagents»
Представляем Вашему вниманию похожие книги на «Polar Organometallic Reagents» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Polar Organometallic Reagents» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.