Catalytic Asymmetric Synthesis
Здесь есть возможность читать онлайн «Catalytic Asymmetric Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Catalytic Asymmetric Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Catalytic Asymmetric Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Catalytic Asymmetric Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Catalytic Asymmetric Synthesis
Catalytic Asymmetric Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Catalytic Asymmetric Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
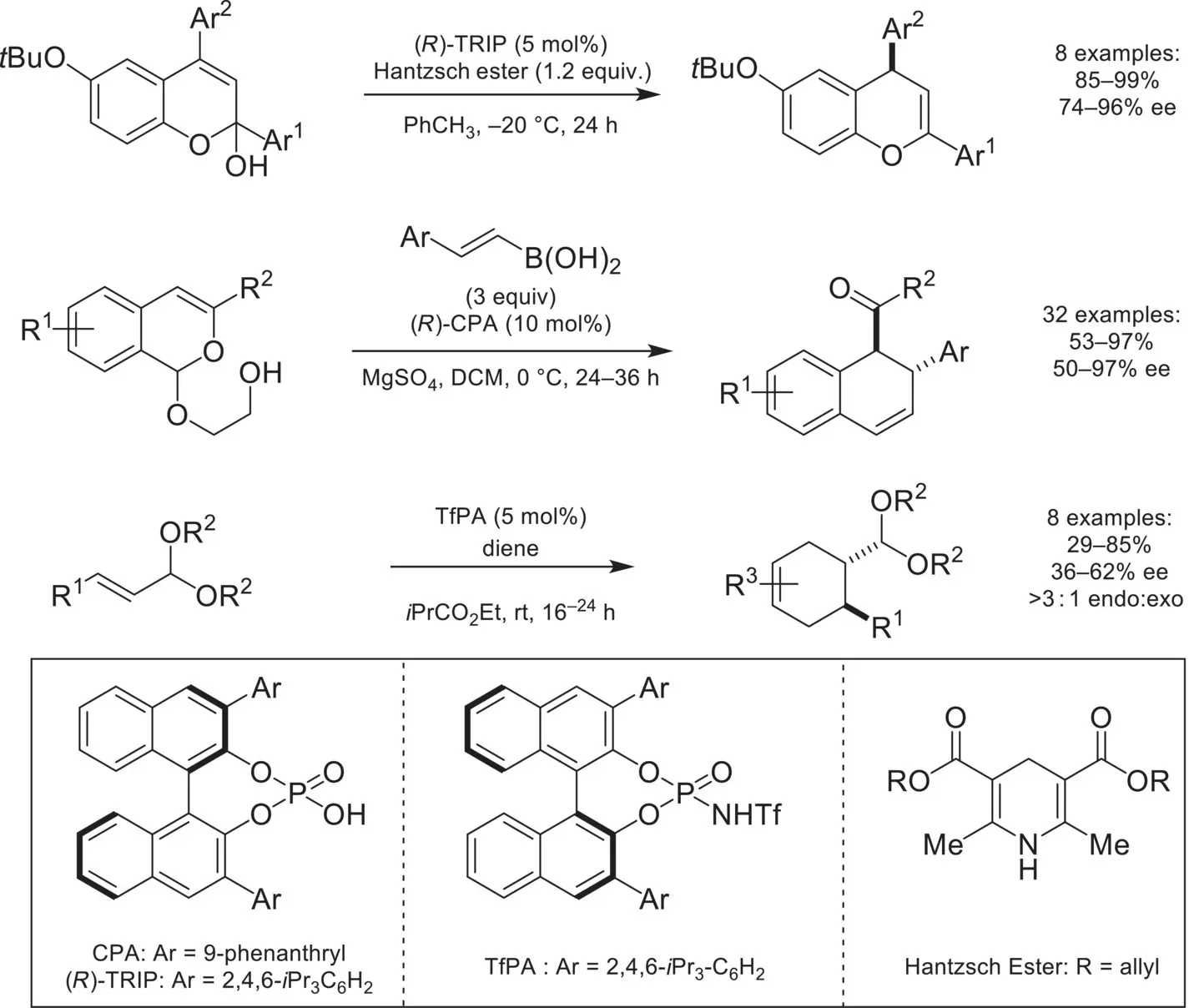
Scheme 4.34. Asymmetric counteranion‐directed catalysis involving stabilized oxocarbenium intermediates.
Source: Based on [115].
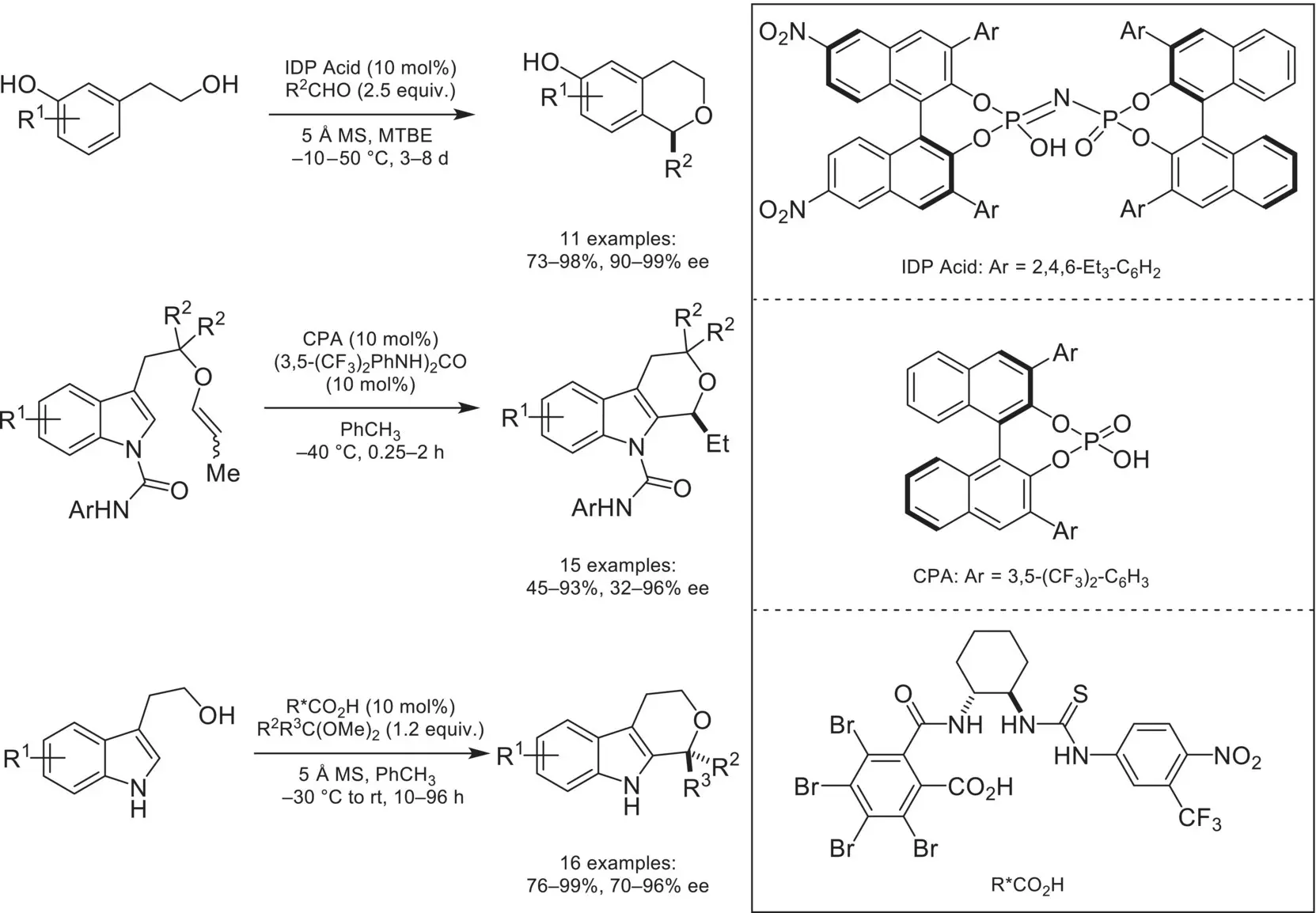
Scheme 4.35. Asymmetric counteranion‐directed catalysis with unstabilized oxocarbenium intermediates.
Source: Based on [118].
Scheme 4.36. Trityl cation/ chiral phosphate salt activation of α‐ketoesters.
Source: Based on [121].
Scheme 4.37. Intramolecular asymmetric counteranion‐directed catalysis with carbocation intermediates.
Source: Based on [122].
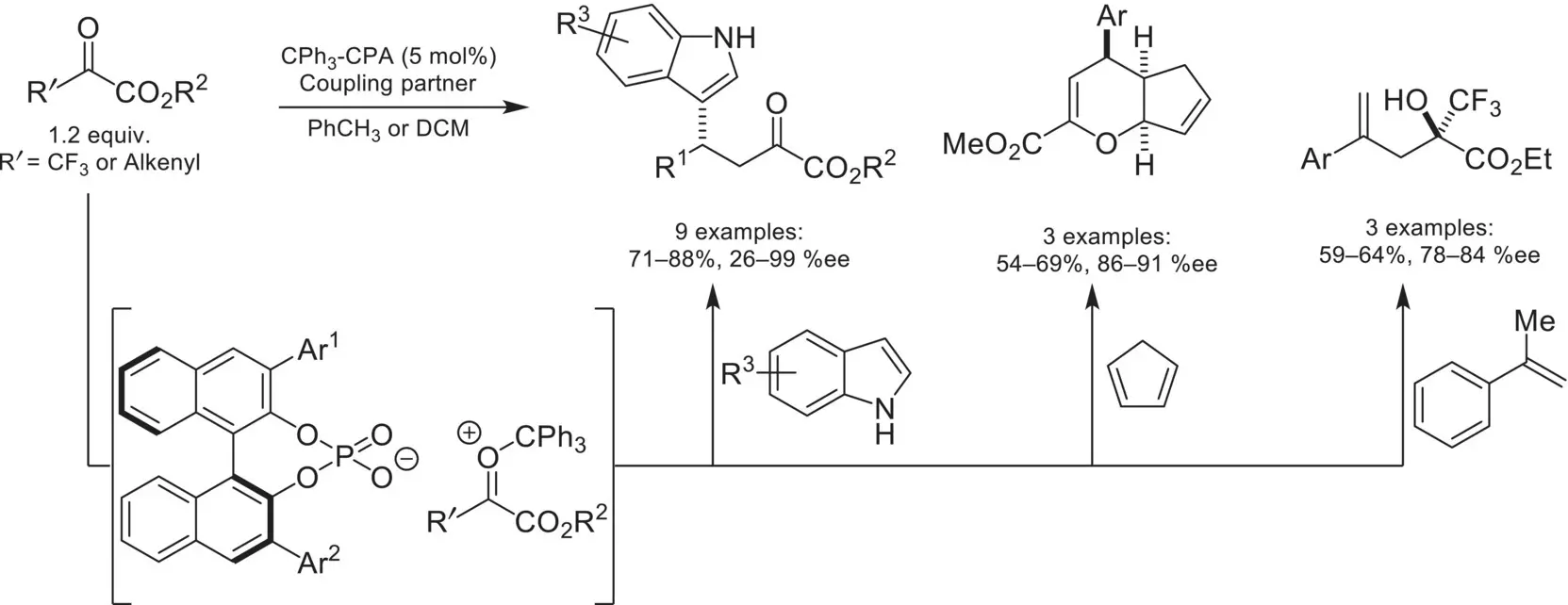
Intermolecular transformations utilizing carbocations in conjunction with chiral‐anions have been of recent synthetic interest. In 2017, the Sun group demonstrated an asymmetric intermolecular reaction involving ion‐pairing with carbocations ( Scheme 4.38) [125]. In this example, a propargyl alcohol is ionized by protonation/dehydration with N ‐triflylphosphoramide catalysts. This intermediate was then shown to undergo nucleophilic attack by 1,3‐diketones and thioacetic acid in high yields and enantioselectivities. Following this, the Peng and Yang groups [126], as well as the Kartika group [127], demonstrated enantioselective intermolecular arylation reactions of allyl alcohols with indoles.
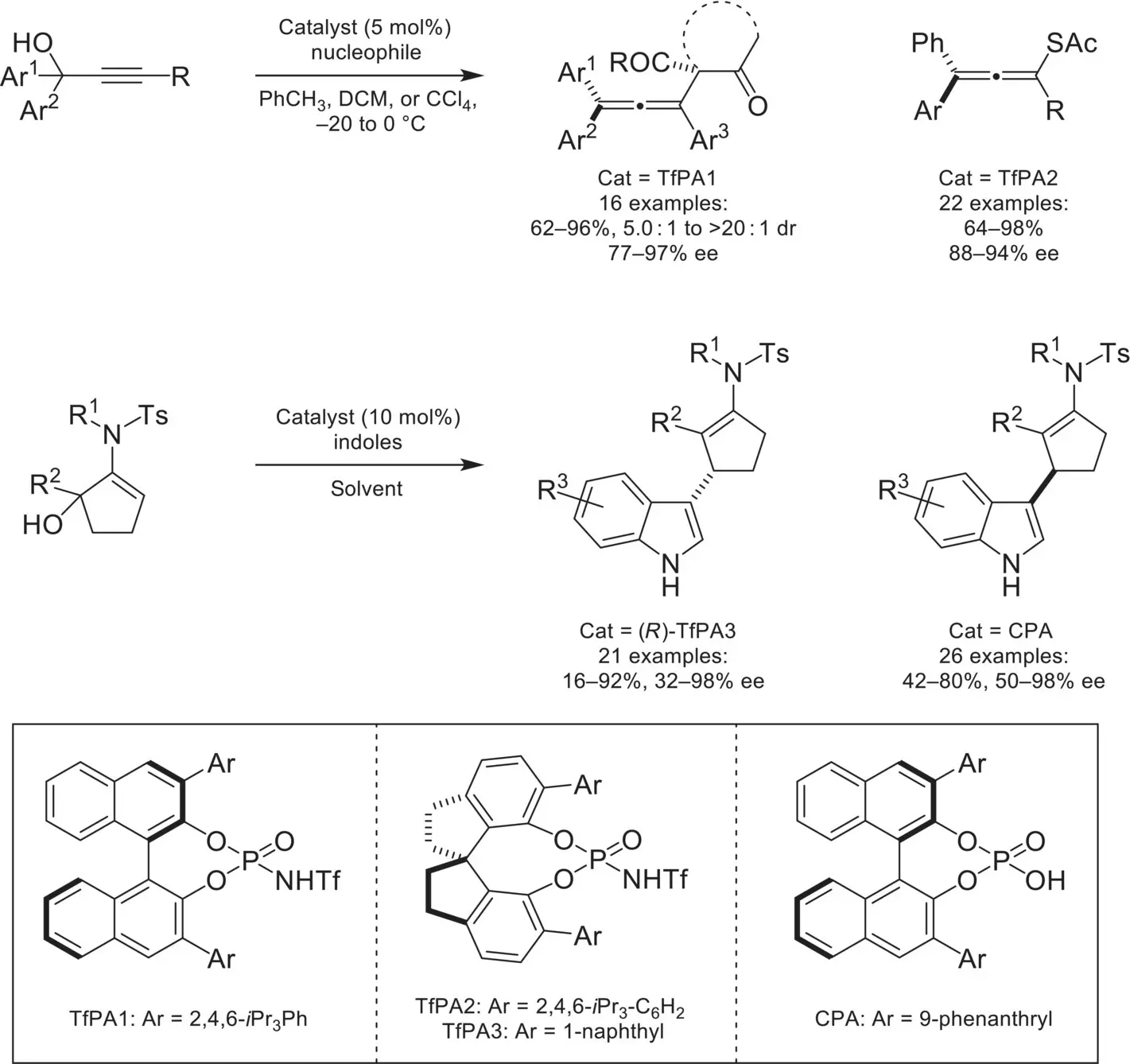
Scheme 4.38. Intermolecular asymmetric counteranion‐directed catalysis with carbocation intermediates.
Source: Based on [125].
4.3.4. Miscellaneous
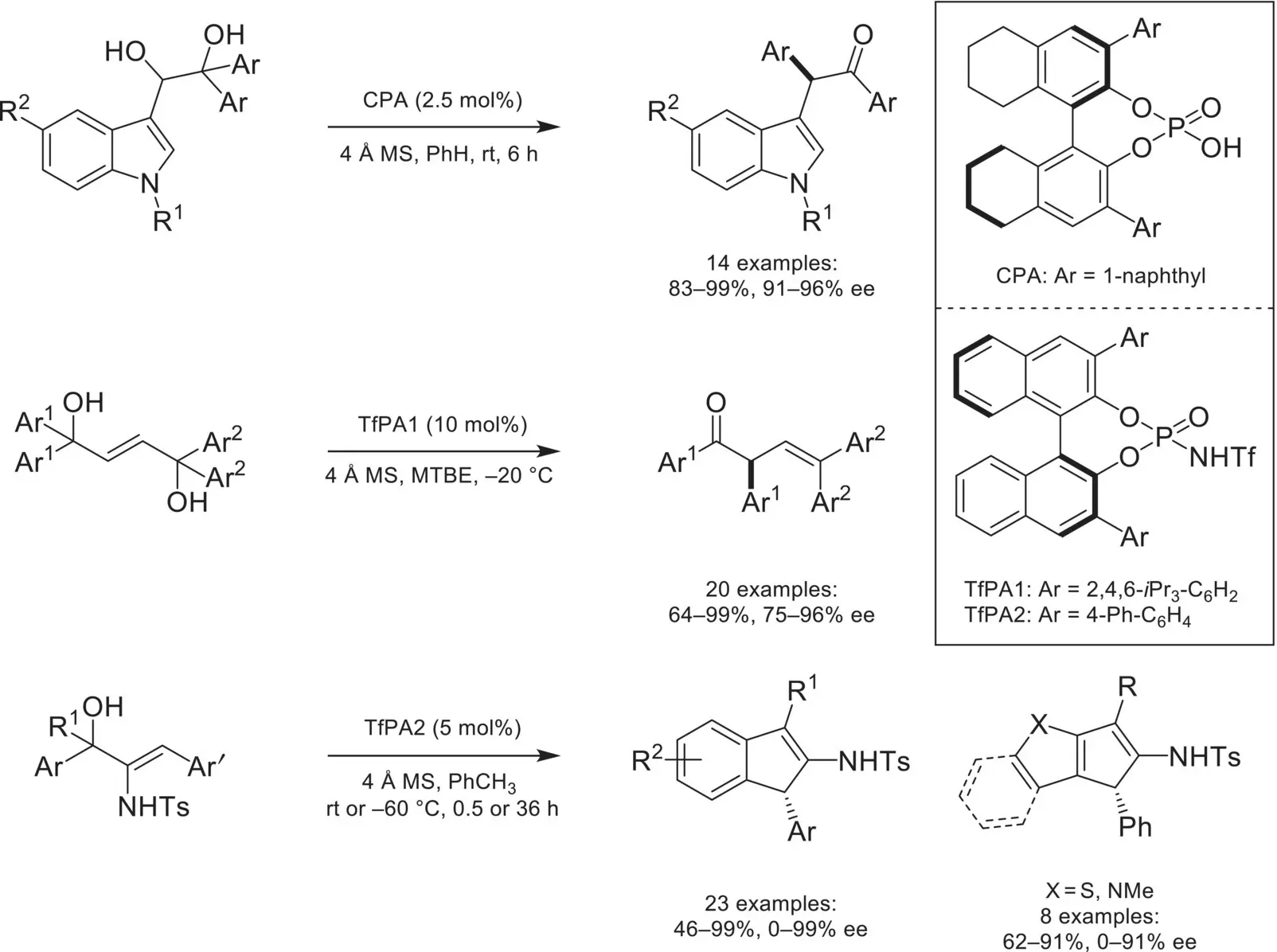
A novel ion‐pairing strategy was demonstrated in 2008 by Toste and coworkers ( Scheme 4.39) [128]. In this example, chiral phosphoric acid or silver phosphate salts would convert amines and sulfides featuring β‐leaving groups into the corresponding aziridinium or episulfonium. The chiral‐anion would cata lyze an asymmetric ring opening of these cationic three‐membered heterocycles with alcohols, to the corresponding 1,2‐amino‐alcohols and β‐alkoxy sulfides in high yields and enantioselectivities. A computational investigation of the mechanism revealed that a C–H•••O interaction between the three‐membered ring’s C–H and the phosphate oxygen is key for stabilizing the ion‐pair intermediate, and is essential for the enantioselectivity of the reaction [129].
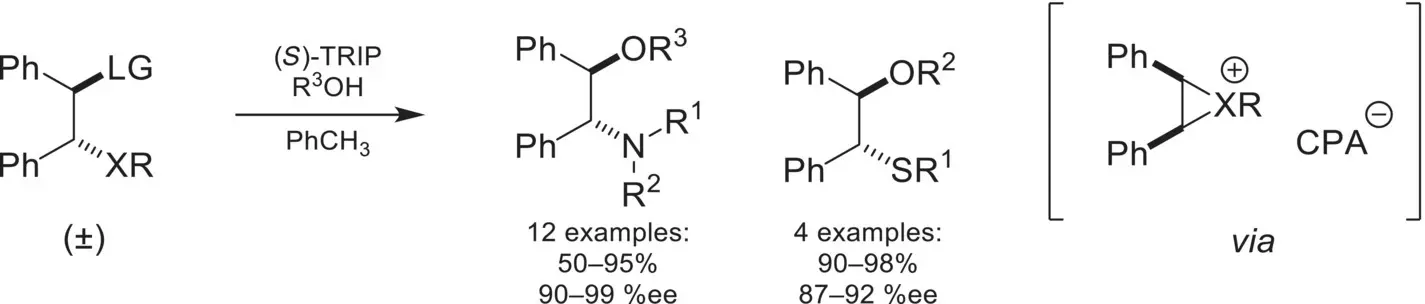
Scheme 4.39. Enantioselective aziridinium and episulfonium ring‐opening.
Source: Based on [128].
Another novel application of chiral‐anion catalysis was demonstrated in 2018 by the Nicewicz group. In this report, a pyrillium/chiral phosphate salt was developed that was catalytically active for a photoredox catalyzed Diels‐Alder, providing bicyclic cyclohexene products in good yield and moderate enantioselectivity ( Scheme 4.40) [130]. Impressively, the reaction could also be conducted intermolecularly, with a cyclic or acyclic diene. This represents a rare example in chiral ion‐pairing catalysis, where the reactivity of radical intermediates is leveraged in an asymmetric fashion.
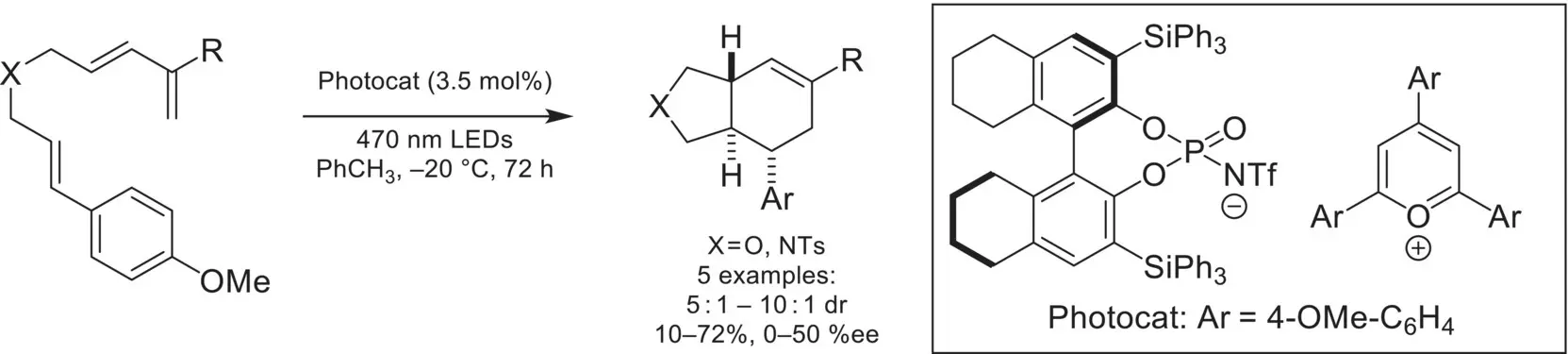
Scheme 4.40. Asymmetric Diels Alder via photoredox/chiral anion dual catalysis.
Source: Based on [130].
4.3.5. Chiral‐Anion Phase Transfer
4.3.5.1. Halogenation
In 2011, the Toste group introduced a novel concept in the field of ion‐pairing catalysis, focused on a chiral‐anion phase transfer (CAPT) strategy ( Scheme 4.41) [131]. The initial report focused on utilizing a catalytically generated SelectFluor/chiral phosphate ion‐pair for the asymmetric fluorocyclization of alkenes. In this system, a deprotonated chiral phosphoric acid catalyst undergoes salt metathesis with insoluble SelectFluor, which introduces a chiral environment around the electrophilic fluorine, allowing it to be transferred with high yields and enantioselectivities to a variety of alkenes. In this system, the phosphate anion serves a dual purpose of activating the substrate via hydrogen bonding and ion‐pairing with the cationic reagent.
In the decade following the initial report of CAPT catalysis, numbers of enantioselective fluorination reactions have been reported with the chiral phosphoric acid/SelectFluor system ( Scheme 4.42). Enantioselective difunctionalization of alkenes constitutes a plurality of this field, and includes enantioselective fluorocyclizations of tryptamines [132], tryptophols [133], and enamides [134], as well as fluorinative semi‐pinacol rearrangement with sulfonamides [135] and alcohols [136]. In addition to difunctionalization reactions, enantioselective α‐fluorinations have also been demonstrated with this system, such as with enamides [137], phenols [138], and branched cyclohexanones [139]. The final class of transformations promoted by this system is enantioselective allylic fluorinations, with amide‐ and phenol‐directing groups [140]. From this, it was later shown that enantioselective fluorination could be accomplished with allylic [141] and homoallylic alcohols [142], utilizing a condensed boronic‐acid‐directing group [143].
Catalysts other than chiral phosphoric acids have also been demonstrated to be compatible with the CAPT strategy. The Hamashima group developed a bifunctional catalyst that mimics the dual functional nature of the chiral phosphate phase‐transfer catalysts ( Scheme 4.43) [144]. This catalyst affected the endo ‐fluorolactonization of 2‐styrenal benzoic acids, where the catalyst’s free alcohol group serves to hydrogen bond with the substrate, while the carboxylate ion‐pairs with the SelectFluor cation. A dimeric version of the catalyst featuring two carboxylates was later developed to synergize with the dicationic nature of SelectFluor. This catalyst was utilized for selective endo ‐fluorocyclization with an amide nucleophile [145], as well as allylic fluorination [146].
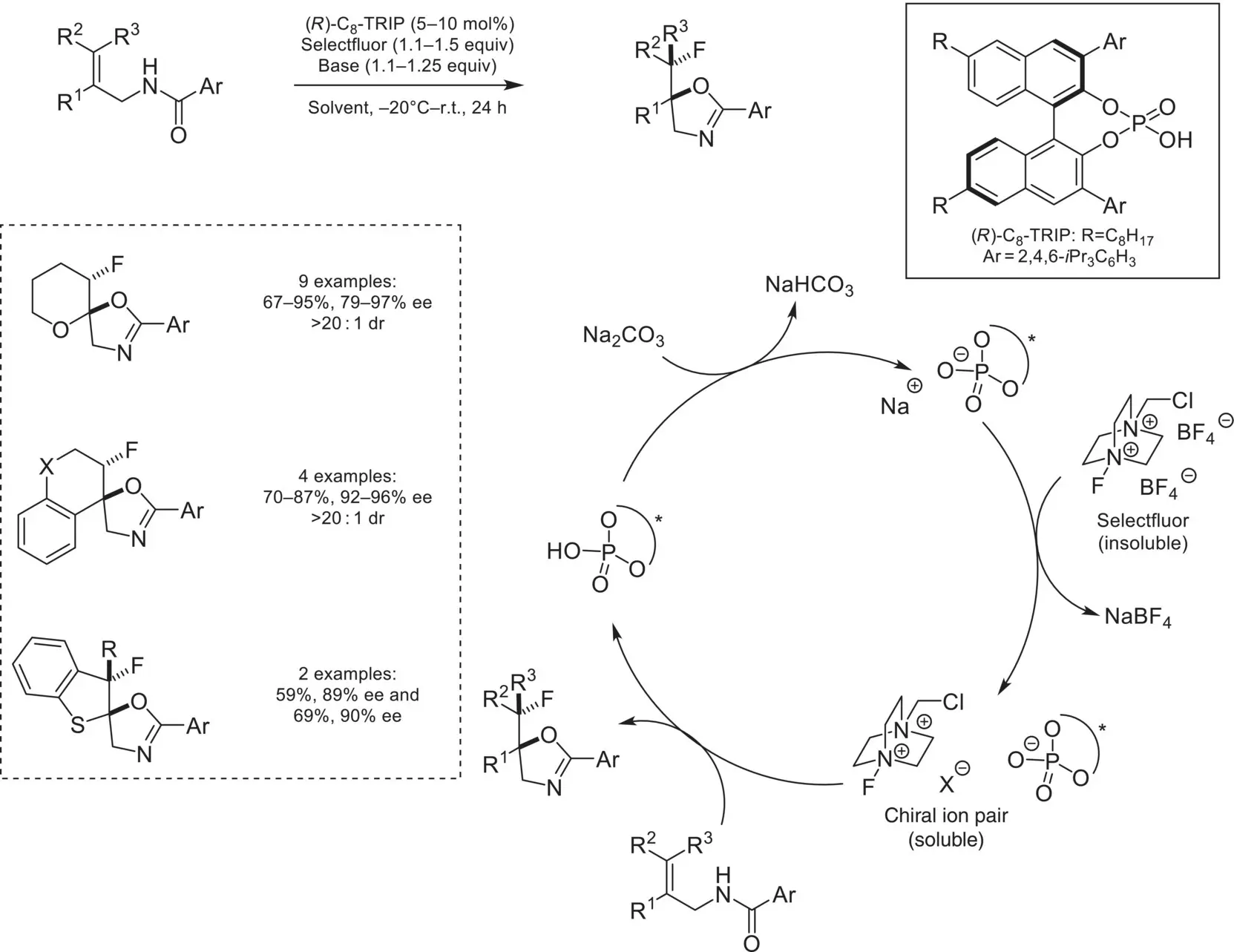
Scheme 4.41. First example of chiral anion phase‐transfer catalysis.
Source: Based on [131].
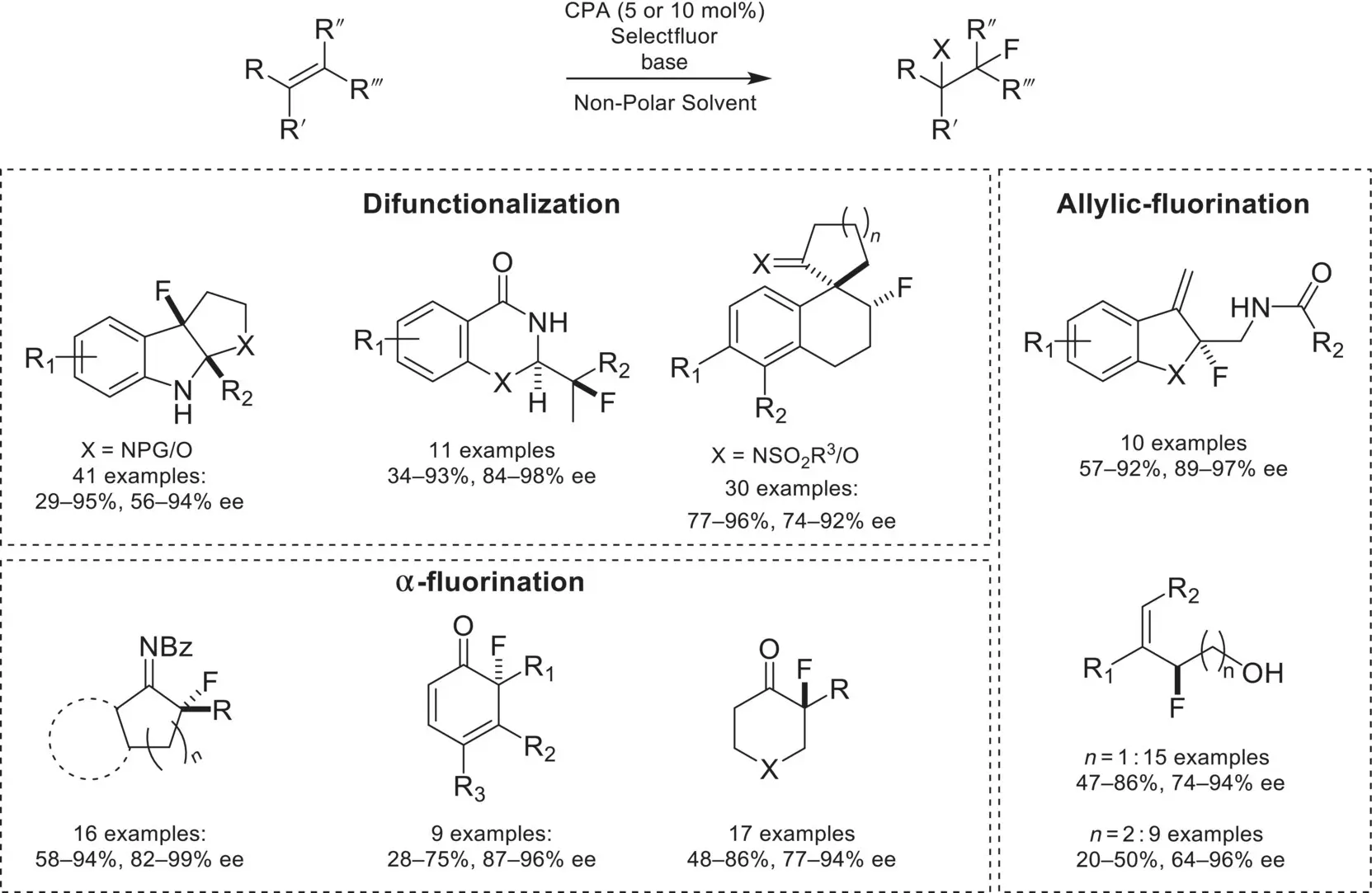
Scheme 4.42. Overview of chiral anion phase‐transfer catalyzed fluorination reactions.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Catalytic Asymmetric Synthesis»
Представляем Вашему вниманию похожие книги на «Catalytic Asymmetric Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.







Обсуждение, отзывы о книге «Catalytic Asymmetric Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.