Catalytic Asymmetric Synthesis
Здесь есть возможность читать онлайн «Catalytic Asymmetric Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Catalytic Asymmetric Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Catalytic Asymmetric Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Catalytic Asymmetric Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Catalytic Asymmetric Synthesis
Catalytic Asymmetric Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Catalytic Asymmetric Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
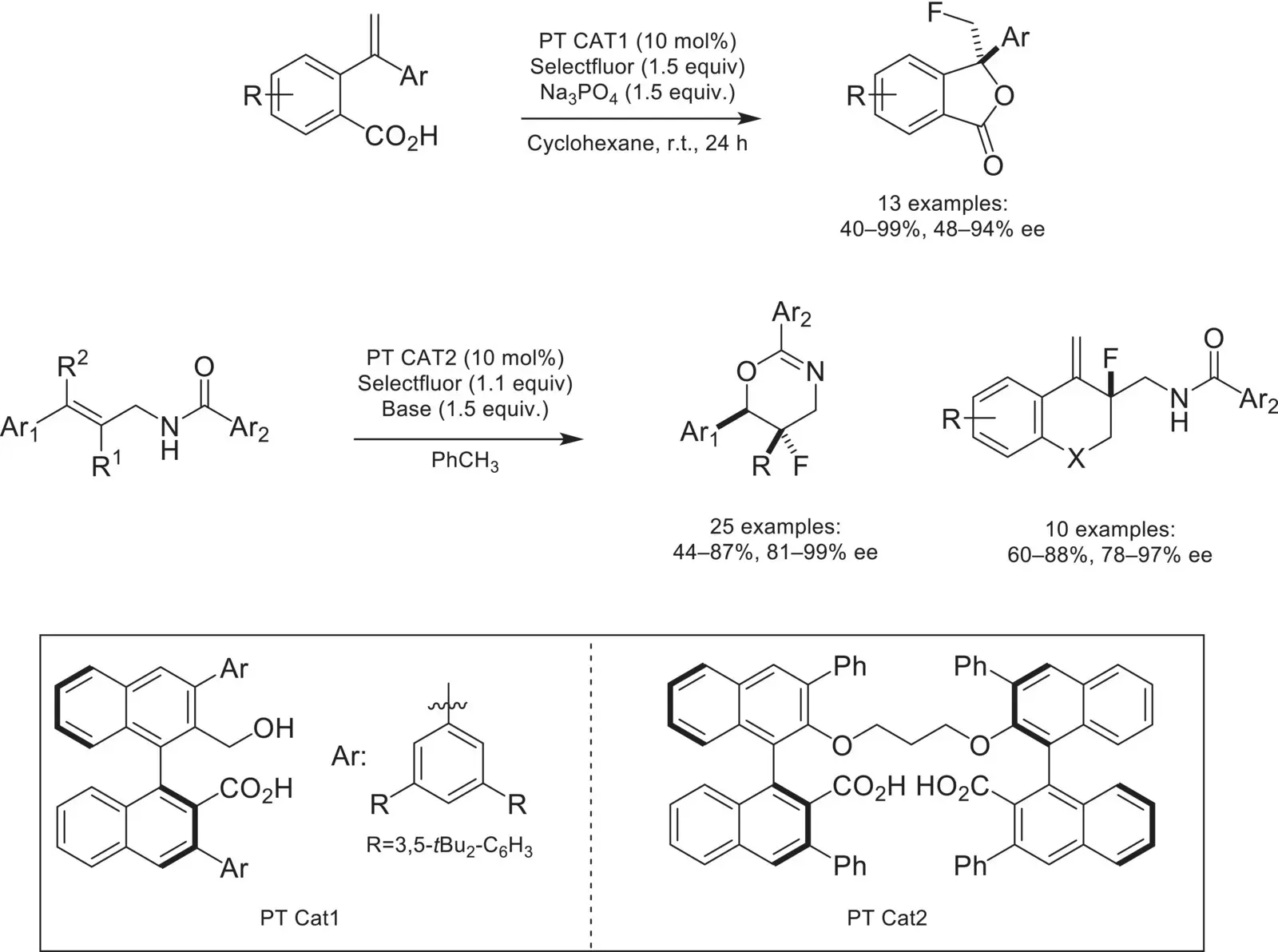
Scheme 4.43. Chiral anion phase‐transfer catalyzed fluorination with BINOL‐derived carboxylate catalysts.
Source: Based on [144].
In addition to fluorination with SelectFluor, CAPT catalysis was adapted to bromo‐ and iodocyclizations by utilizing DABCOnium‐based halogenating reagents ( Scheme 4.44) [147]. This was first demonstrated by the Toste group with halocyclization of alkenes with tailored‐reagents based on the SelectFluor scaffold. This strategy was found to be applicable to bromocyclization of difluoroalkenes, providing access to tetrasubstituted CF 2Br‐containing stereocenters [148]. These compounds were derivatized to a variety of pharmaceutically relevant difluoromethylene‐containing moieties, such as difluoromethyl, difluoroester, and difluorophosphonate. The DABCOnium‐based brominating reagents have been applied to the enantioselective bromocyclization of other classes of substrates, such as tryptamines [149], tryptophols [150], and indenes [151]. Similarly to asymmetric fluorination, the CAPT bromination and iodination strategies were applicable to reactions outside of halocyclizations, such as halogenative semi‐pinacol rearrangements [136] as well as atroposelective bromination [152].
4.3.5.2. Amination
Due to the generality of the CAPT strategy, other cationic electrophiles could be deemed asymmetric by amending them to a phase‐transfer setting. In 2014, the Toste group demonstrated that aryl diazonium salts were suitable electrophiles for CAPT catalysis, by developing an asymmetric diazenation of tryptamines ( Scheme 4.45) [153]. In a 2015 follow‐up publication, the strategy was applied to carbonyl‐containing nucleophiles to generate quaternary diazenated stereocenters [154].
4.3.5.3. Miscellaneous Transformations
In 2013, the Toste group demonstrated the compatibility of oxopiperidinium salts in conjunction with CAPT catalysis for an enantioselective cross‐dehydrogenative coupling reaction ( Scheme 4.46) [155]. Instrumental to this was the development of novel triazole‐containing phosphoric acids, where it was hypothesized that these were engaging in noncovalent interactions with the substrate, leading to high enantioselectivities. A follow‐up mechanistic study focusing on a data‐intensive approach revealed that a π‐stacking interaction between the triazole and the substrate was the primary factor in controlling enantioselectivity [156]. The oxopiperidinium/chiral‐anion system was later utilized in a deracemization reaction of indolines via oxidation to the 3‐H indole scaffold proceeding, and subsequent reduction to the indoline [157]. In 2016, the Toste group reported the asymmetric arylation of benzopyrylium with phenols under phase‐transfer conditions ( Scheme 4.47) [158].
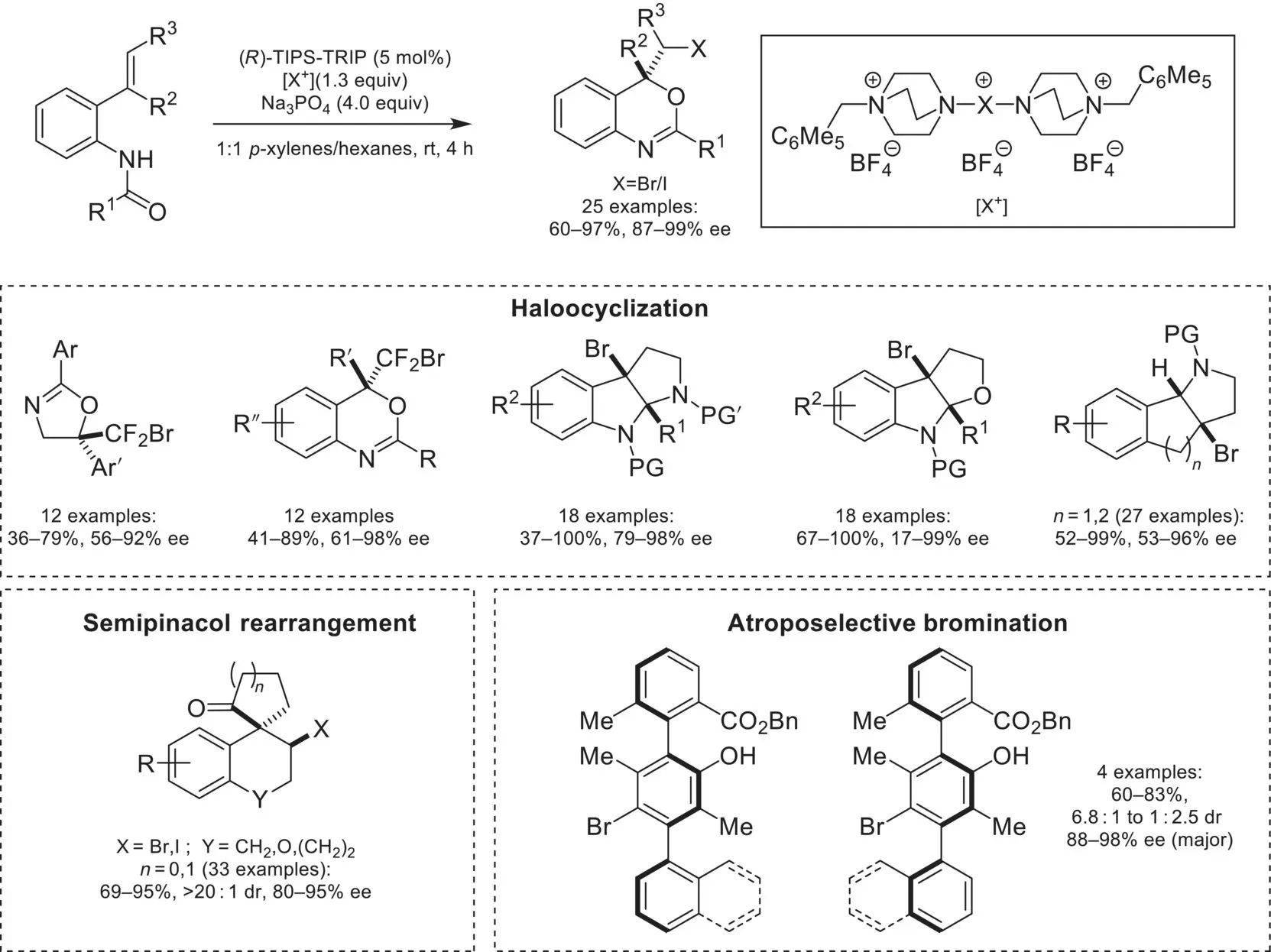
Scheme 4.44. Overview of chiral anion phase‐transfer catalyzed bromination and iodination.
Source: Based on [147].
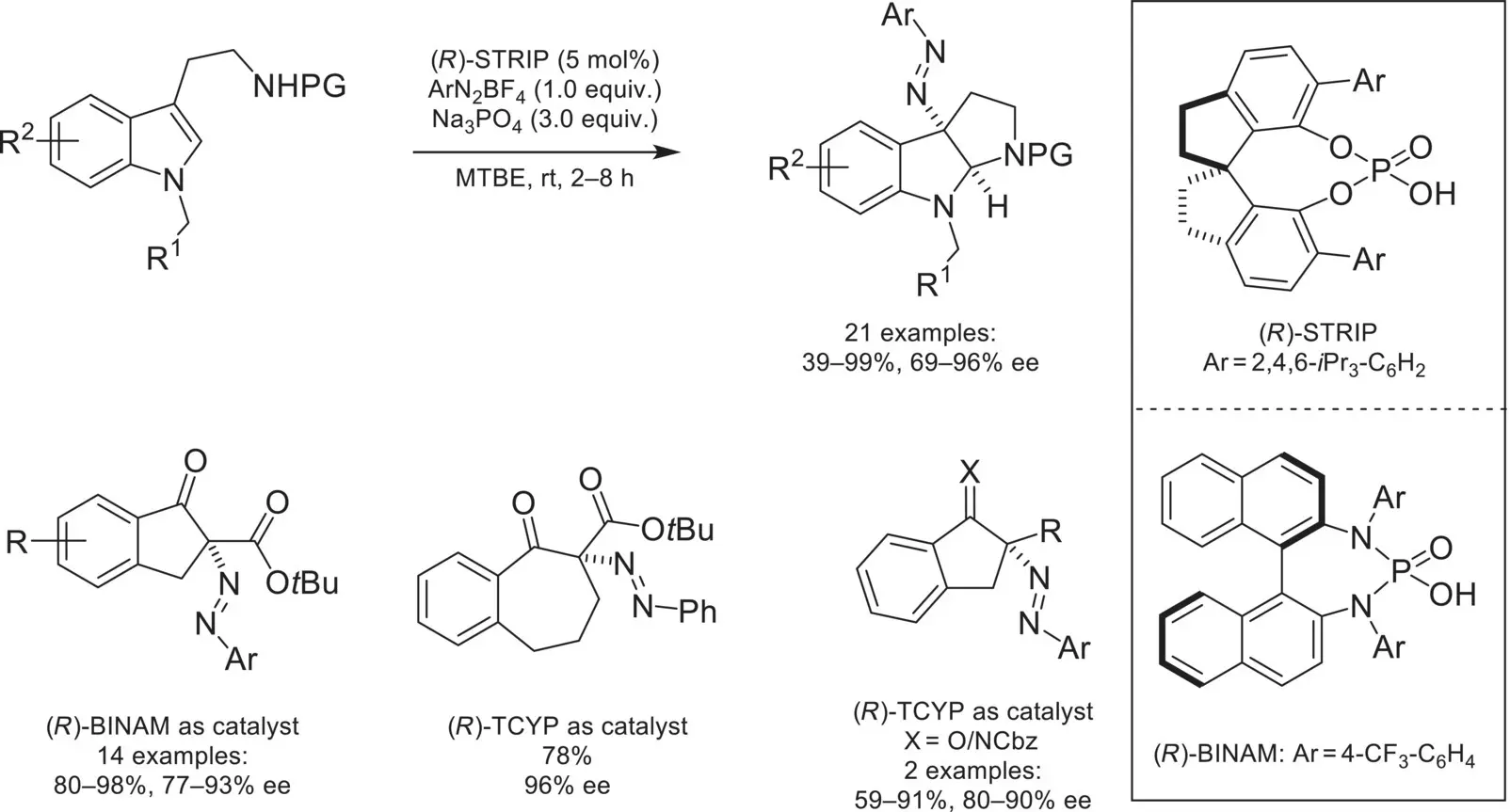
Scheme 4.45. Chiral anion phase‐transfer catalyzed diazenation.
Source: Based on [153].
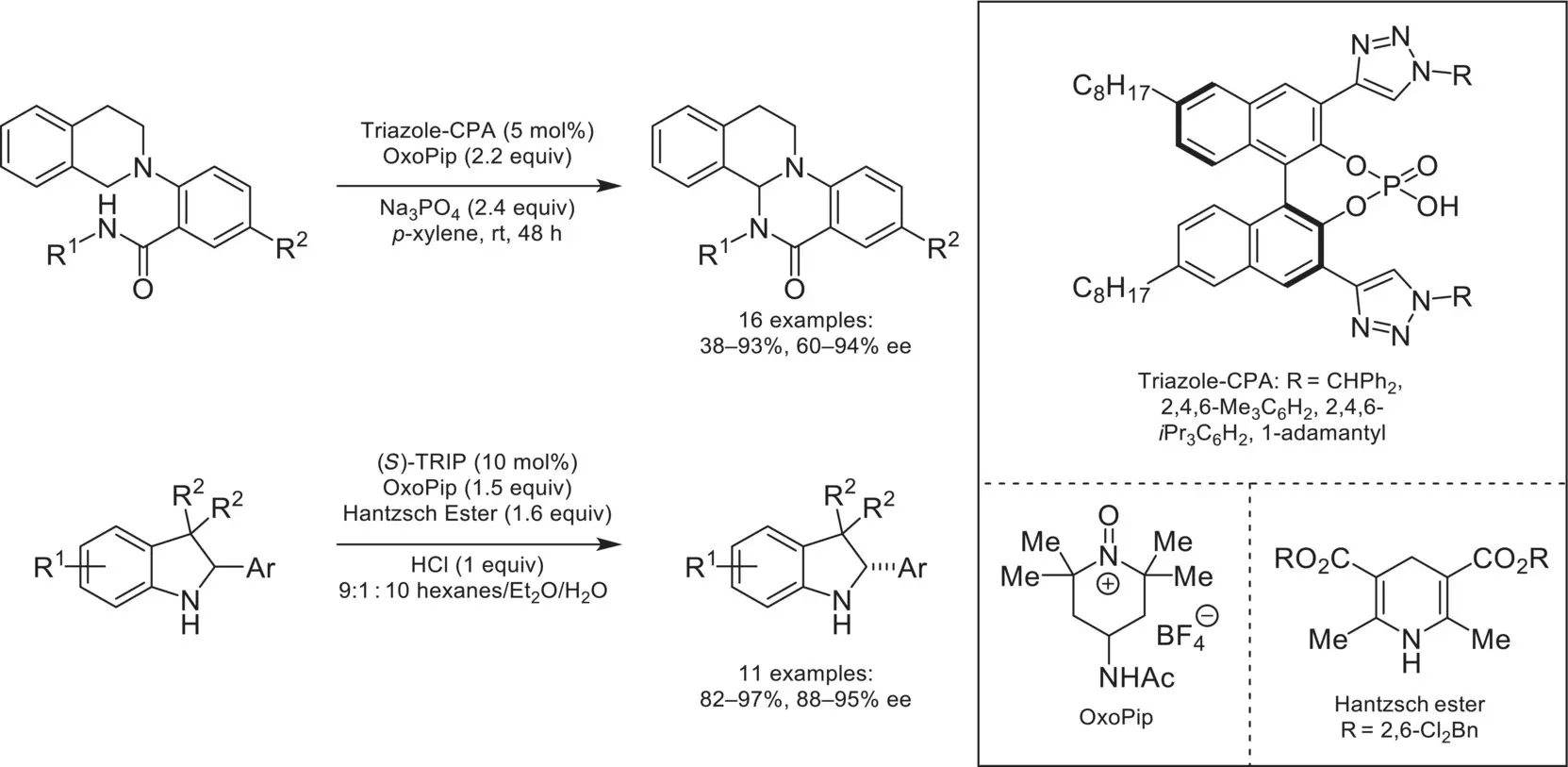
Scheme 4.46. Asymmetric transformations mediated by oxopiperidinium/chiral anion salts.
Source: Based on [155].
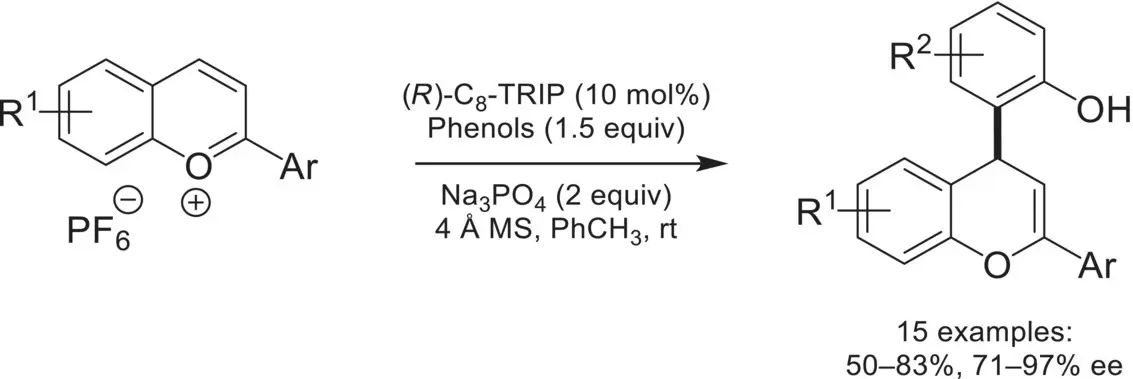
Scheme 4.47. Asymmetric arylation of benzopyrylium with phenols under phase‐transfer conditions.
Source: Based on [158].
4.3.6. Transition‐Metal/Chiral‐Anion Dual Catalysis
With the prevalence of cationic transition‐metal complexes in catalysis, there was a strong interest in rendering these reactions asymmetric by utilizing ion‐pairing with chiral counterions. In 2007, the Toste group demonstrated this principle with an enantioselective Au(I) catalyzed hydro‐cyclization of allenes ( Scheme 4.48) [159]. The chiral‐anion strategy was crucial for success in this system, as the linearity of Au(I) complexes rendered attempts to use chiral ligands for enantioselectivity unsuccessful. By combining an achiral gold complex with a chiral phosphate counter anion, the enantioselective cyclization of alcohols, sulfonamides, and carboxylic acids could be achieved in high yields. Solvent choice was found to be a key factor for achieving selectivity, as more polar solvents resulted in weaker ion‐pairing, leading to diminished selectivities. This strategy was later adapted to the desymmetrization of 1,3‐diols via intramolecular cyclization of allenes [160]. In the succeeding decade, the robustness of this strategy was demonstrated by the compatibility of chiral‐anions with a wide variety of transition‐metal‐catalyzed reactions.
The combination of Pd catalysis with chiral‐anions has led to many asymmetric methodologies. Of particular initial interest was the generation of cationic Pd‐allyl complexes with a chiral counteranion for asymmetric Tsuji–Trost‐type allylations. This was first demonstrated by the List group in 2007, with an asymmetric α‐allylation of aldehydes ( Scheme 4.49) [161]. An intramolecular version of this reaction was later reported by the Toste and Sigman labs, where pyrrolidines and benzomorpholines were accessed in high yields and enantioselectivities [162]. In addition to ion‐pairing with a cationic transition‐metal center, the Ooi lab demonstrated that enantioselectivity can be achieved by utilizing an achiral cationic ligand ion‐paired with a BINOL‐derived anion [163]. In addition to Pd(II) allyl complexes, the Toste group demonstrated an enantioselective 1,1‐arylborylation of alkenes, which proceeds through an enantiodetermining migratory insertion, followed by β‐hydride elimination and reinsertion [164]. By changing the coupling partner to aryl boronic acids, an enantioselective 1,1‐diarylation was achieved [165]. Additionally, exclusion of a coupling partner was found to result in an asymmetric Heck–Matsuda arylation, generating cyclic arylated stereocenters in high yield and enantioselectivity [166].
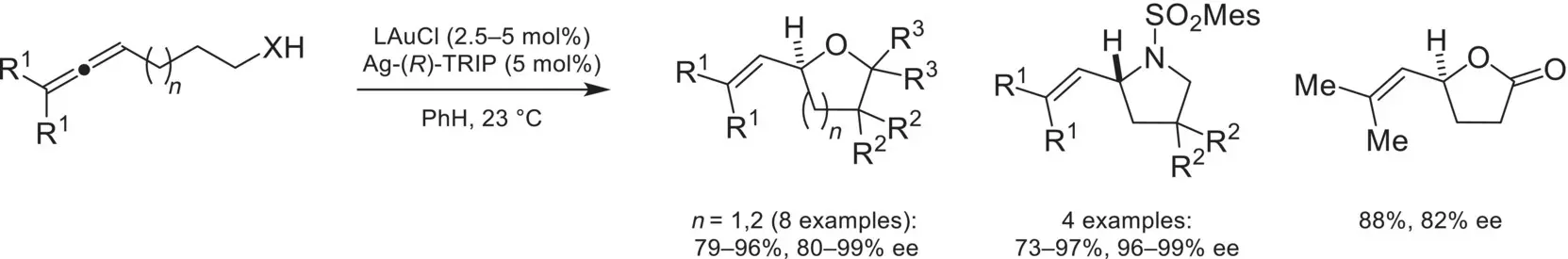
Scheme 4.48. First example of a transition‐metal/chiral anion catalyzed transformation.
Source: Based on [159].
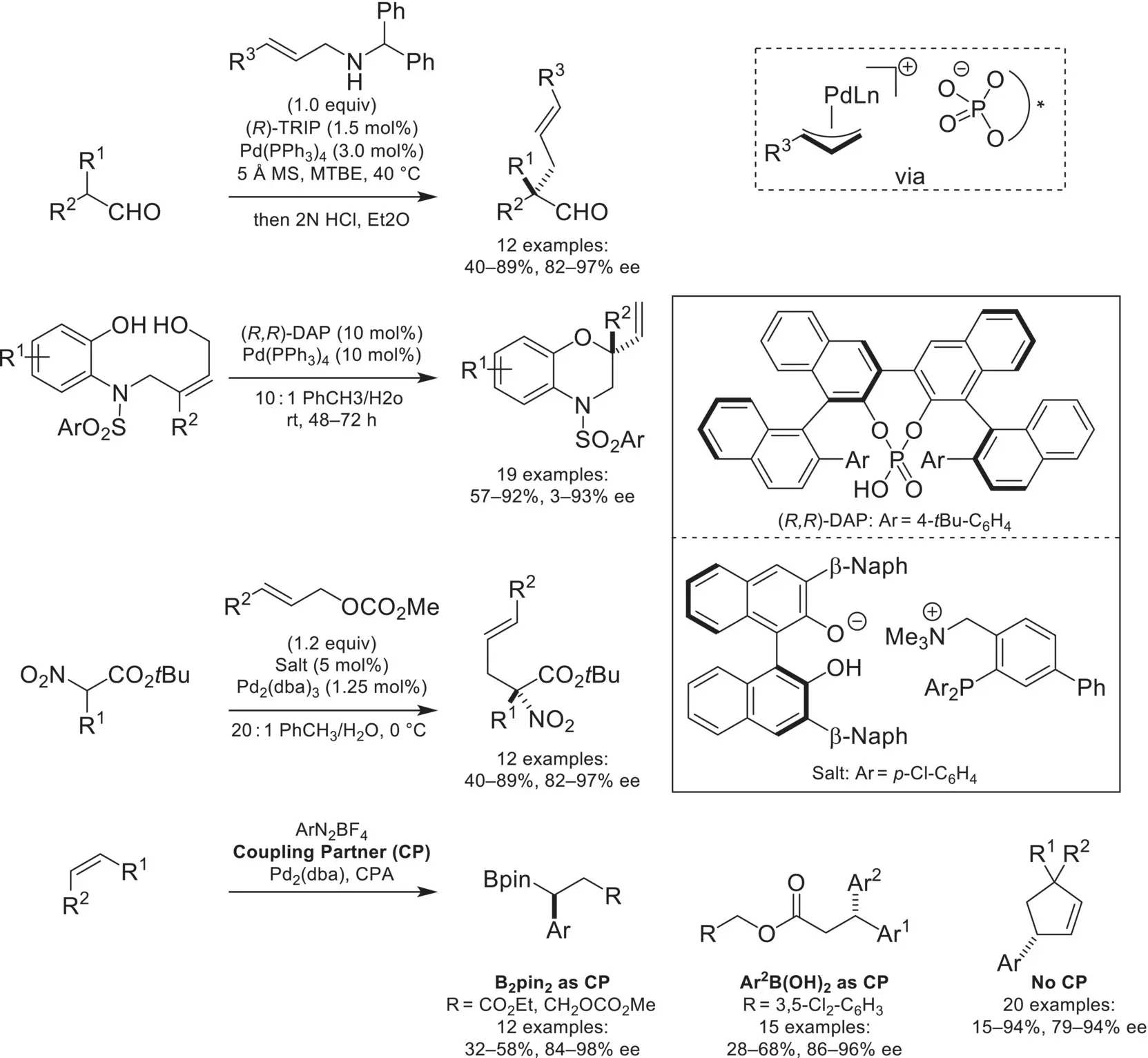
Scheme 4.49. Asymmetric transformations catalyzed by a Pd/chiral phosphate ion pair.
Source: Based on [161].
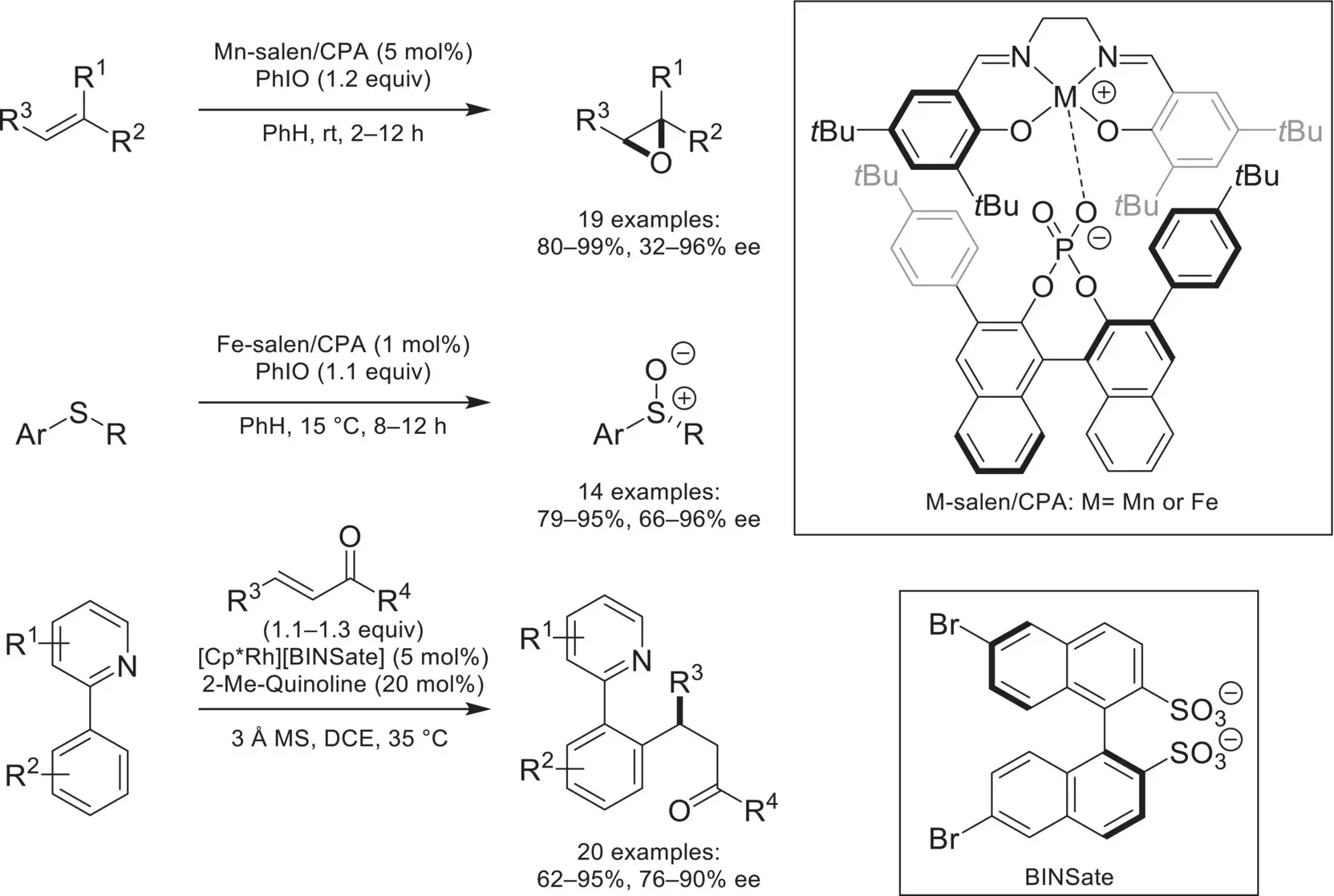
Scheme 4.50. Asymmetric transformations catalyzed by other transition‐metals ion‐paired with chiral anions.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Catalytic Asymmetric Synthesis»
Представляем Вашему вниманию похожие книги на «Catalytic Asymmetric Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.







Обсуждение, отзывы о книге «Catalytic Asymmetric Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.