Systematics and the Exploration of Life
Здесь есть возможность читать онлайн «Systematics and the Exploration of Life» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Systematics and the Exploration of Life
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Systematics and the Exploration of Life: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Systematics and the Exploration of Life»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Systematics and the Exploration of Life — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Systematics and the Exploration of Life», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Proteins can be roughly divided into four classes according to their morphology: globular proteins, which are in an aqueous environment, fibrous proteins, which form large aggregates and mostly constitute the cytoskeleton, membrane proteins and so-called “disordered” proteins, which are generally small and have no inherent fixed structure. Here, we will only discuss the first category of proteins, which are the globulars.
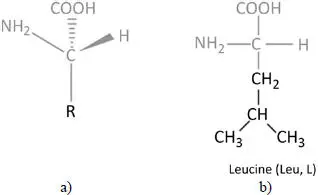
Figure 2.1. a) General structure of an amino acid; b) chemical formula of leucine
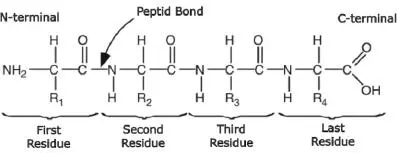
Figure 2.2. A polypeptide consisting of four amino acids (tetrapeptide)
2.2. Folding
Proteins adopt a specific spatial organization, most often called a “structure”. This structure is crucial for their function. This relationship between structure and function, established by Emil Fisher at the end of the 19th century, is the foundation of structural biology. Methods used for determining the structure of proteins have evolved considerably, but the method of choice remains as X-ray diffraction, which requires the protein to be crystallized. The data bank listing protein structures (as well as nucleic acids and some sugars) is the PDB (Protein Data Bank) (Berman et al . 2000). The number of resolved protein structures is growing rapidly from year to year. A protein could theoretically adopt a large number of three-dimensional conformations, but most of them spontaneously fold into a particular and unique stable form. This particular shape is due to the fact that the peptide backbone groups and side chains interact with each other and with water. Thus, some conformations have more stabilizing interactions than others and are therefore favored (Alberts et al . 1994). The paradigm of the relationship between the protein sequence and its three-dimensional (3D) structure comes from Christian Anfinsen’s studies on ribonuclease (Anfinsen 1973). Anfinsen showed that proteins isolated in solution can regain their original active conformation after denaturation. Therefore, the conclusion was that all the information needed to fold a protein must be inherent to its amino acid order (Alberts et al . 1994). Other studies have also drawn the same conclusions, leading to the general theory that the amino acid sequence of a protein specifies its conformation (Stryer 1994).
“Water-soluble” proteins fold into a compact globular form (unlike fibrous, membrane and “disordered” proteins). The hydrophobic nature of certain amino acids makes this compact folding necessary. Indeed, the side chains of the non-polar residues are hydrophobic and are grouped together within the globular structure of the protein – isolated from the surrounding water – while the polar residues, and the polar groups of the backbone, are hydrophilic and form hydrogen bonds with water or with each other. This spatial distribution of hydrophobicity has been known since the middle of the 20th century, in particular thanks to the work of Bressler and Talmud (1944), who described a globular protein as a micelle, with a predominantly hydrophobic core surrounded by a predominantly hydrophilic crown. The hydrophobic and hydrogen bonds are largely responsible for the stability of the protein structure, obtained under the effect of the only available energy source, thermal agitation. These continuous fluctuations lead to more or less rapid displacements around stable local conformations, called secondary structures. Alpha helices (30% of the residues) and beta strands (20% of the residues) are the most frequent. These local structures are stabilized by hydrogen bonds due to the particular values of the dihedral angles of the peptide backbone.
2.3. Substitution(s) in protein structures
The substitution of one amino acid by another (a mutation) is one of the fundamental events of molecular evolution, with variable consequences in proteins. The majority of mutations have no major effect on the phenotype – they are neutral – but some of them can cause disease (Studer et al . 2013). Indeed, local changes may occur in binding sites with other molecules and can thus affect the function of proteins (Gong et al . 2009), but long-term effects on the overall structure can also be observed (Zhou et al . 2007).
Many studies agree that the majority of substitutions have no significant effect on the overall structure, stability or function of the protein. As a matter of fact, it has been shown that 75% of the amino acids can be modified without significant alteration of the protein structure (Sander and Schneider 1991; Shakhnovich and Gutin 1991; Schaefer and Rost 2012). These observations support the neutral hypothesis of point mutations, but it is important to keep in mind that this does not mean that all mutations are neutral: the majority of point mutations are effectively counter-selected because their impact is negative for the cell. For example, the probability that a human DNA repair enzyme, 3-methyladenine DNA glycosylase, becomes non-functional after a random mutation is 34% (± 6%), and this proportion can be extended to other families (Guo et al . 2004).
2.4. Effect on overall structure and function
From a structural point of view, the effect of an amino acid substitution is difficult to model because the effects can be drastic. A striking example is the L16A mutation (leucine 16 replaced by alanine) of a DNA-binding protein of Drosophila melanogaster , located in the homeodomain, which profoundly modifies the structure of the native protein while maintaining the three helices (Religa et al . 2005) ( Figure 2.3).
However, the link between the structural change and functionality, or disease is not obvious or systematic. For example, in the case of human lysozymes with many known structures, two natural mutants, D67H and I56T, form amyloid fibrils in the extracellular space of multiple organs and tissues, resulting in non-neuropathic systemic amyloidosis. In the case of the D67H mutation ( Figure 2.4(a)), the structure of the lysozyme is highly disrupted in two of its loops, while the structure of the I56T mutant ( Figure 2.4(b)) appears little disrupted, as shown by the structures of the mutants superimposed on that of the native lysozyme. Nevertheless, without attempting to predict whether a mutation will be pathogenic or not, the modeling of the effect of a mutation on a protein has been proposed by assessing the variation in its stability.
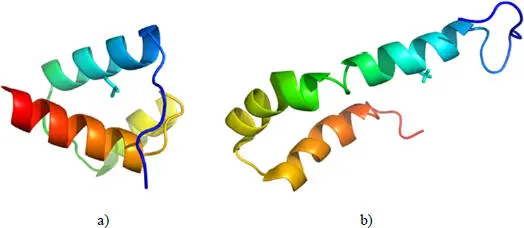
Figure 2.3. a) DNA binding protein of Drosophila melanogaster (PDB code 1enh); b) L16A mutant of the same protein (PDB code 1ztr). The mutated position is shown with its side chain. For a color version of this figure, see www.iste.co.uk/grandcolas/systematics.zip
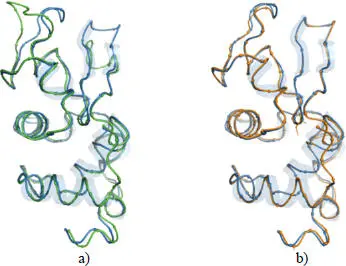
Figure 2.4. a) Superimposed structures of the D67H mutant (green, PDB code 1lyy) and the native structure (blue, PDB code 2nwd) of the human lysozyme; b) superimposed structures of the I56T mutant (orange, PDB code 1loz) and the native structure (blue, PDB code 2nwd) of the human lysozyme. The mutated positions are represented with their side chains. For a color version of this figure, see www.iste.co.uk/grandcolas/systematics.zip
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Systematics and the Exploration of Life»
Представляем Вашему вниманию похожие книги на «Systematics and the Exploration of Life» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Systematics and the Exploration of Life» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.