Systematics and the Exploration of Life
Здесь есть возможность читать онлайн «Systematics and the Exploration of Life» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Systematics and the Exploration of Life
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Systematics and the Exploration of Life: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Systematics and the Exploration of Life»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Systematics and the Exploration of Life — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Systematics and the Exploration of Life», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
2.5. Effect on stability
Proteins are said to be “marginally stable”: typically, there is a difference of 3–7 kcal/mol in free folding energy (ΔG) between folded and unfolded conformations. Amino acid side chain substitutions thus have a significant effect on protein stability: the effect of a single mutation is on average −0.95 kcal/mol according to the Protherm database (Gromiha and Sarai 2010), which in 2017 included the measurements from ΔΔG for 1,866 proteins with their structure. ΔΔG is the difference between the free folding energy of the native and the mutated protein.
This low protein stability is assumed to be either the result of a balance between function and stability (DePristo et al . 2005), or the result of a balance between destabilizing mutations and highly unstable proteins (Taverna and Goldstein 2002; Bloom et al . 2007; Zeldovich et al . 2007).
Two principles must be kept in mind:
– function is often dependent on a dynamic effect of structure;
– any protein must be degradable at a cost that is not restrictive for the cell.
Many algorithms and web servers have been developed to provide an estimate of the variation of Gibbs’ free energy (ΔΔG) under the effect of a point mutation: FoldX (Guerois et al . 2002) and Rosetta (Kellogg et al . 2011) are among the best known. SPROUTS (Lonquety et al . 2009) is a web server combining the results of several methods. These methods try to predict whether a given mutation will be destabilizing, neutral or stabilizing. The comparison between predicted and experimental energy variations gives globally satisfactory results ( Figure 2.5(a)): the correlation between the predictions of FoldX and the measurements of Protherm is 0.59. Nevertheless, the difference can be quite large in some cases (Lonquety et al . 2009). It is interesting to note that the prediction results are quite different when comparing wild to mutant and mutant to wild, in absolute value ( Figure 2.5(b)).
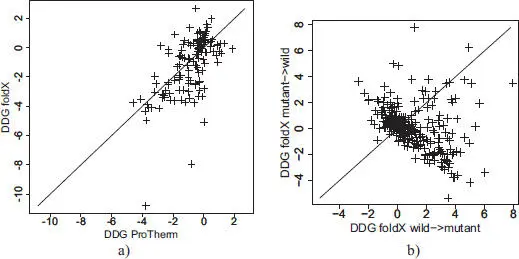
Figure 2.5. Comparison of predicted ΔΔG by FoldX and those experimentally measured (Protherm)
COMMENT ON FIGURE 2.5.– a) Prediction by FoldX of ΔΔG for 130 proteins present in Protherm and belonging to the 11 families in which at least 20 different point mutant structures are known (see section 2.6 for a description of this dataset). b) Prediction by FoldX of ΔΔG for the families in which at least 20 different point mutant structures are known. The abscissa shows the predicted value of ΔΔG from the native to the mutant, and the ordinate shows the predicted value of ΔΔG from the mutant to the native.
2.6. Effect on the peptide backbone
In the calculation methods of ΔΔG mentioned above, the main chain is fixed; only the side chains are allowed to move. The range of motion of the side chain is extracted from the rotamer libraries (Dunbrack 2002), and their degrees of freedom strongly depend on the conformation of the main chain. Taking the deformation of the main chain into account should improve our understanding of the consequences of substitution. Nevertheless, it is legitimate to ask whether the backbone is really disrupted by a simple substitution, at least in the one we can observe, and whether this disruption is measurable with current techniques. Nowadays, the resolution of the structure of proteins determined by crystallography and X-ray diffraction is of the order 1 Å, which means that the precision of the structures obtained is of the order 1/10 Å (Luzzati 1952). The displacement of the backbone atoms caused by substitution is typically also 1/10 Å, which puts us at the limit of resolution.
In addition, protein structures of identical sequences can vary greatly due to different protein–protein interactions, interactions with different ligands or solvents (Kosloff and Kolodny 2008). However, provided that a sufficient number of structures are available, the effect of the mutation can be distinguished from “noise”, in other words, from statistical fluctuations due to other sources, such as exposure to solvents or belonging to a secondary structure (Shanthirabalan et al . 2018). This requires measuring the variations between the native and mutated proteins, and considering both the global variability and local flexibility of the structure.
The objective is therefore to measure whether a given structure (generally the mutant structure) is different at certain places (locally) – for example, at the place of mutation – from another so-called “reference” structure (generally the native structure). These two proteins have extremely close sequences (for a punctual mutation), so it is easy to find the alignment (correspondence) between the amino acids of the two proteins. Then, the two structures (in green and red, Figure 2.6(a)) are superimposed to minimize the distances between the corresponding pairs of Cα. We will then measure the distances between the Cαs of the two structures for small fragments (in blue, Figure 2.6(a)) of three consecutive Cαs by calculating the RMSD (Root-Mean-Square Deviation). A length of three residuals was chosen because it is the length for which the measured local effect is the strongest (Shanthirabalan et al . 2018). This calculation is performed for all residues and we obtain a “profile” of the RMSDs ( Figure 2.6(b), graph) for a given protein pair.
In order to determine whether RMSDs tend to be larger in terms of mutations, it is necessary to be able to compare several structures that only differ from each other by a single point mutation. In PDB, there are 11 families with at least 20 mutants with a single substitution from a reference structure, for a total of 580 mutants and 11 reference structures (Shanthirabalan et al . 2018). When comparing the profiles between proteins of the same family, it can be observed that the regions with the largest RMSDs are often the same, regardless of the position of the mutation. Overall, some proteins “move” very little, while others “move” a lot ( Figure 2.7). It is therefore difficult to know whether a large RMSD is due to the mutation, to the intrinsic flexibility of the protein or to an overall variability resulting from the various crystallization conditions. Thus, if a comparison is made between the distribution of RMSDs centered on mutated residues and the distribution of RMSDs for all positions of the structures of the 11 families, one will observe that the two distributions significantly overlap ( Figure 2.8(a)). To better differentiate the effect due to mutations, the two other sources of variability must be neutralized.
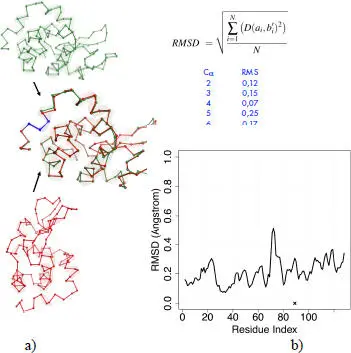
Figure 2.6. Procedure for calculating “local” RMSDs. For a color version of this figure, see www.iste.co.uk/grandcolas/systematics.zip
COMMENT ON FIGURE 2.6.– a) Superimposition of two lysozyme structures. b) Their difference is measured by the RMSD calculated for the fragments of three successive residues, such as the fragment in blue. The RMSD is the root of the sum of the N distances D between the alpha carbon pairs ai and b’i (i.e. bi after superimposition) divided by the N number of C α pairs (three in our case). This calculation is performed on the whole protein. There are as many RMSDs as residues in the protein (except for the two C α s at the extremities); a profile is obtained (graph below). The mutation is localized at the location of the cross (protein 2hef chain A, mutation I89A).
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Systematics and the Exploration of Life»
Представляем Вашему вниманию похожие книги на «Systematics and the Exploration of Life» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Systematics and the Exploration of Life» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.