Solar-to-Chemical Conversion
Здесь есть возможность читать онлайн «Solar-to-Chemical Conversion» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Solar-to-Chemical Conversion
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Solar-to-Chemical Conversion: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Solar-to-Chemical Conversion»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Solar-to-Chemical Conversion — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Solar-to-Chemical Conversion», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
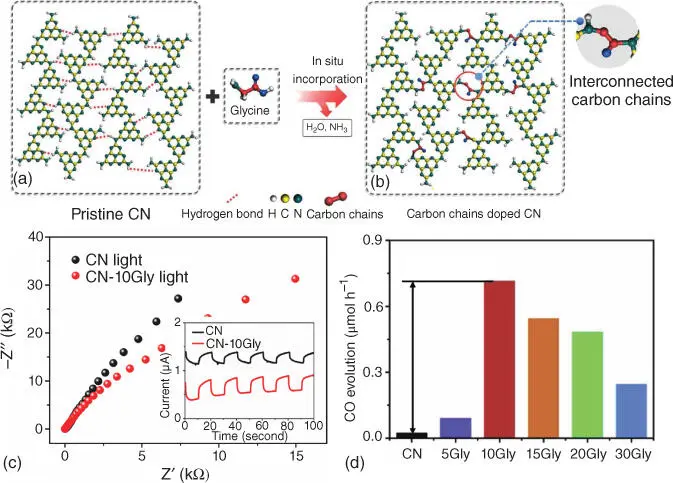
Figure 2.13Structures of CN before (a) and after (b) doping with carbon chains. (c) Electrochemical impedance spectroscopy (EIS) Nyquist plots under irradiation condition. Inset: Periodic on/off photocurrent response under visible‐light irradiation. (d) Comparison of the photocatalytic CO 2reduction rate of the samples with different amount of glycine, respectively.
Source: Ren et al. [70].
On the other hand, it is reported that CO can originate from the secondary photolysis of unstable reduced products, such as HCO 2H. For example, Frei and coworker investigated the reaction mechanism of CO 2photoreduction over Ti silicalite molecular sieve in the presence of methanol as electron donor by means of in situ FTIR [73]. It was found that HCO 2H, CO, and HCO 2CH 3as reduced products were detected, in which mass proportion of CO was the highest. The formation of the products was studied through the infrared analysis of experiments with isotope‐labeled reactants, such as C 18O 2, 13CO 2, and 13CH 3OH. The results inferred that the produced CO is derived from secondary photolysis of the reduced HCO 2H. In contrast, the formic acid is the primary two‐electron reduction product of CO 2at the ligand‐to‐metal charge transfer transition (LMCT) excited Ti centers. This means that since the complex hydrocarbons are the target products, the photolysis effect should be paid more attention for suppressing the formation of CO.
2.4.3 Dioxygen (O 2)
As discussed above, an ideal artificial photosynthesis system usually contains at least three different components: light harvesting, water oxidation, and CO 2reduction [74]. To achieve CO 2photoreduction accompanied with water oxidation, early single integrated systems are composed of semiconducting metal oxide with large band gaps absorbing UV light and metal or metal oxide cocatalysts, where considerable efforts are paid to functionalizing these materials and exploring various modifications [75]. Nevertheless, since the assembly of multifunctional units in an integrated device is extremely difficult, an alternative strategy is to divide the overall process into two half‐reactions: water oxidation and CO 2reduction. Once each half‐reaction is well understood and optimized, the two reactions can be coupled in an integrated device. For example, it is demonstrated that combining two semiconductor materials in tandem (Z‐scheme) mode can efficiently extend the light response into the visible region [76]. Recently, Arai et al. reported a photoelectrochemical system consisting of SrTiO 3photoanode for water oxidation and InP photocathode for CO 2reduction that produced O 2and formate, respectively [77]. Besides, mononuclear [Ru(bpy) 3] 2+is often combined with S 2O 8 2−sacrificial acceptor to evaluate water oxidation photocatalysis systems in aqueous photosensitization system in the past decades [78]. For example, upon combining mild oxidant [Ru(bpy)] 3 3+of well‐defined potential (+1.26 V) with a Co 3O 4nanotube in neutral aqueous solution, a hole can be efficiently transferred to the catalyst [79]. Subsequently, four holes transfer from [Ru(bpy) 3] 3+to Co 3O 4and react with two H 2O molecules to O 2and 4H +, which indicates that Co 3O 4is appropriate candidate for use in an artificial photosynthetic assembly.
On the water oxidation side, several groups proposed approaches to overcome challenges of efficiently combining molecular light absorbers with multi‐electron water oxidation catalyst inspired by the tyrosine mediator design of nature's photosystem II. For instance, the Mallouk group reported a series of researches on coupling of Ir oxide nanocluster (IrO x) with [Ru(bpy) 3] 2+or a porphyrin visible‐light absorber through a benzimidazole–phenol redox linking with both components covalently anchored on a TiO 2surface [80]. Based on the results of transient optical spectroscopy, when the Ru chromophore absorbs a visible photon, an photoinduced electron injects into TiO 2at an ultrafast speed, and subsequently an electron transfers from the benzimidazole–phenol mediator to the oxidized Ru complex for reducing the oxidized chromophore on a short time scale compared to the catalytic turnover of O 2at the IrO xparticle. As a result, the competition between undesired transfer of electrons injected into TiO 2back to the oxidized light absorber and hole injection into the IrO xcatalyst is shifted in favor of catalysis, thus improving the quantum efficiency of water oxidation by a factor of three. The approach offers substantial room for further efficiency improvement because the relative position of the light absorber and the catalyst with the attached mediator is not yet molecularly defined in the present system.
Recently, earth‐abundant inorganic oxide catalysts have been widely used in water oxidation, accompanied with CO 2photoreduction. In the past decades, Ir oxide nanoclusters have been viewed as efficient catalysts for water oxidation, which can be used to assemble directly various fashion systems for closing the photosynthetic cycle on the nanoscale. For example, Kim et al. prepared an all‐inorganic polynuclear unit consisting of an oxo‐bridged binuclear ZrOCo IIgroup with iridium oxide nanocluster assembled on SBA‐15 silica mesopore surface, which could produce CO and O 2simultaneously [81]. Apart from Ir‐based cocatalyst, other noble metals also are applied into the CO 2photoreduction and water oxidation by deposition on semiconducting materials. For example, Fan et al. fabricated a binary functional photocatalyst by loading Au@Pd nanoparticles onto oxygen vacancy‐rich TiO 2through surfactant‐free deposition–reduction method [82]. The obtained V O‐rich TiO 2contributes more protons to water oxidation process, and meanwhile, the metallic Au@Pd sites are beneficial for CO 2activation and proton supply. By varying the ratio of Au@Pd nanoparticles and V Oconcentration, CH 4selectivity of 96% can be obtained with minimal H 2production. However, the Ir oxide or other noble metals are not a viable component for a scalable artificial photosystem because of the scarcity of this element. Therefore, earth‐abundant metal oxides are developed as robust alternatives. For example, Yu et al. Cl‐doped Cu 2O nanorods for photocatalytic CO 2reduction conjugated with H 2O oxidation under visible‐light irradiation [83]. Owing to the introduction of Cl atoms, the band structure of Cu 2O is optimized, leading to a more positive VB position for water oxidation, which promotes CO 2activation and separation and transfer efficiency of photoinduced charge carriers. As a result, in Figure 2.14, the Cl‐doped Cu 2O composite exhibits excellent photocatalytic CO 2reduction performance accompanied by favorable water oxidation ability under visible‐light irradiation. DFT calculations demonstrate that the doping of Cl atoms facilitates to transform CO 2into the intermediates of *COOH, *CO, and *CH 3O, which could enhance production of reductive CO and CH 4and oxidative O 2. The CO, CH 4, and O 2amount were improved to 1.74 μmol cm −2, 0.39 μmol cm −2, and 1.32 μmol cm −2, respectively, for the Cu 2O–Cl‐4, as shown in Figure 2.14a. Furthermore, it is found that the Cl‐doped Cu 2O composite displays stronger affinity toward the *CO intermediate, which tends to be protonated and ultimately produce CH 4, leading to higher selectivity of CH 4than that of pure Cu 2O.
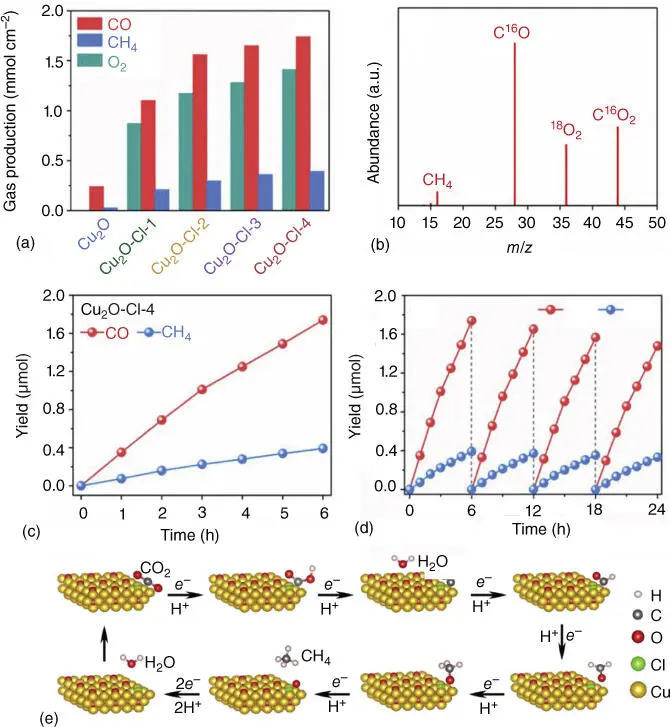
Figure 2.14(a) Gas production under six hours visible‐light irradiation of pure Cu 2O and Cl‐doped Cu 2O samples. (b) Mass spectrum from the 18O‐labeling H 2O isotope experiments of Cu 2O–Cl‐4. (c) Time‐dependent product evolution over Cu 2O–Cl‐4 under visible‐light irradiation. (d) Stability test of Cu 2O–Cl‐4. (e) Proposed reaction pathways of CO 2RR to CO and CH 4on Cl‐doped Cu 2O.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Solar-to-Chemical Conversion»
Представляем Вашему вниманию похожие книги на «Solar-to-Chemical Conversion» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.

![Евгений Матерёв - Музеи… или вдохновляющая музыка The Chemical Brothers [litres самиздат]](/books/437288/evgenij-materev-muzei-ili-vdohnovlyayuchaya-muzyka-th-thumb.webp)










Обсуждение, отзывы о книге «Solar-to-Chemical Conversion» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.