Biofuel Cells
Здесь есть возможность читать онлайн «Biofuel Cells» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Biofuel Cells
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Biofuel Cells: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Biofuel Cells»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
This book covers the most recent developments and offers a detailed overview of fundamentals, principles, mechanisms, properties, optimizing parameters, analytical characterization tools, various types of biofuel cells, all-category of materials, catalysts, engineering architectures, implantable biofuel cells, applications and novel innovations and challenges in this sector. This book is a reference guide for anyone working in the areas of energy and the environment.
Biofuel Cells — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Biofuel Cells», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Glucose can be oxidized by a variety of enzymes including glucose oxidase and glucose dehydrogenase. Of these, glucose oxidase has been the one massively preferred, due to its high specificity and good turnover and stability [14, 15]. Glucose oxidase (EC 1.1.3.4) is a dimeric flavoprotein that oxidizes β-D-glucose into D-glucono-δ-lactone while reducing molecular dioxygen (from here on simply referred to as oxygen) to hydrogen peroxide. In each subunit, an active site is deeply buried in a funnel-shaped pocket that contains a non-covalently bound flavin adenine dinucleotide (FAD) cofactor ( Figure 1.1a). The N5 atom of this molecule is situated 13–18 Å from the surface and acts as the first electron acceptor in a so called “ping-pong” mechanism [16]. This first half reaction (enzyme reduction) takes place through simultaneous donation of a proton and a hydride from the glucose to the His516 residue and FAD, respectively. Although literature frequently states that the product of this half-reaction is FADH 2, there is evidence of the resulting negative charge in the flavin moiety. Therefore, the reduced state of the cofactor is better described as the anionic form FADH −[17]. The second half-reaction is the reoxidation of FADH −to FAD, reducing an oxygen molecule to peroxide. This last process takes place in two one-electron steps that produces two intermediates, a semiquinone radical for the FAD flavine moiety and a superoxide anion radical for the O 2molecule ( Figure 1.1b). Although not directly participating in the electron transfer, it is believed that His559 and Glu412 help with the pH control in the active site.
Glucose oxidase is produced by a variety of animals, plants, bacteria, algae and fungi. However, only GOx extracted from this last kingdom (mainly from Aspergillus and Penicillium genera) have gained industrial application, partly because they fall under the “generally recognized as safe” category of the U.S. Food and Drug Administration [14]. In academic fuel cell research, GOx produced by Aspergillus niger is highly preferred mainly due to its commercial availability. A few studies have been reported using GOx from Penicillium funiculosum 46.1 but the enzyme extraction and purification from the cell culture needs to be performed [13].
Although efficient, it is thought that wild-type glucose oxidase is not at its catalytic maximum, and therefore directed evolution experiments have been performed to find better versions of the enzyme. In a recent study, Petrović et al . found several GOx mutants that presented a higher rate for glucose oxidation reaction, as well as a smaller Michaelis-Menten constant ( K M). Molecular dynamics simulations and X-ray crystallographic information revealed that a key mutation was the exchange of Met556 for a valine residue modifying the shape of the active site. In wild-type GOx, the His516 residue can have two conformations, a catalytic and an inactive one, in which its imidazole side chain flips into a cavity near the active site ( Figure 1.2a). When Met556 was substituted for a valine, the cavity became smaller, effectively locking the His516 into the catalytic position [15]. This is reflected in the relative values of the calculated free energies (Δ G ) for the catalytic and non-catalytic states. While in wild-type GOx both states have similar Δ G , the mutated enzyme clearly shows thermodynamic and kinetic preference for the catalytic state ( Figure 1.2c). This example shows that deeper understanding of the mechanistic subtleties can have convenient implications in the industrial applications that employ GOx, including fuel cells.
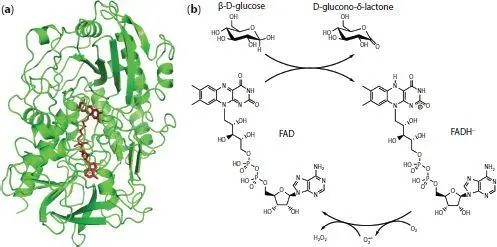
Figure 1.1 (a) Structure of a subunit of glucose oxidase from Aspergillus niger elucidated by Hecht et al . [18] (PDB code 1GAL), showing the FAD cofactor buried in the protein. (b) Catalytic reaction of glucose oxidase. The semiquinone radical FAD intermediate is not shown for clarity.
Laccase (EC 1.10.3.2) is a multicopper “blue” oxidase that has a variety of organic and inorganic compounds as its natural substrates, including phenols, phosphates, acids, ketones and amines. In all cases, O 2acts as the final electron acceptor. It is formed by a single polypeptide chain that folds into three different beta barrel domains. Its active site contains four copper atoms, classified as T1, T2, T3α and T3β, according to their spectroscopic and paramagnetic properties. T1 Cu, for example, presents an intense blue color because of its coordination with nitrogen and sulfur atoms from the neighboring histidine and cysteine residues. On the other hand, T2 Cu presents similar absorption to aquo or hydroxo Cu complexes [19]. In the enzyme oxidized state, all the Cu atoms have an oxidation state of +2.
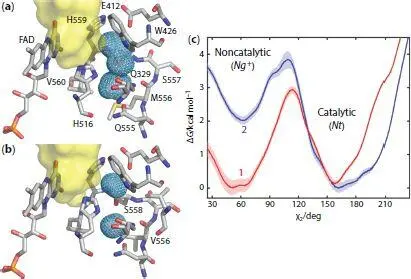
Figure 1.2 Molecular dynamics studies of the structure of the active site of wild-type (a) and mutant (b) glucose oxidase. The meshed area in the right-hand side is a pocket in which His516 can flip into. In the mutant form, the pocket is reduced in size and divided in two parts, effectively locking His516 out. (c) Comparison of the calculated free energy for the catalytic and non-catalytic states in wild-type (curve 1) and mutant (curve 2) GOx. Republished with permission, from ACS Catal ., Dušan Petrović et al ., 7, 2017, 6188–6197 available online at https://pubs.acs.org/doi/10.1021/acscatal.7b01575; further permissions related to the material excerpted should be directed to the ACS.
The T1 Cu is located in a substrate binding pocket close to the enzyme surface and is the initial electron acceptor in a one-electron oxidation of the substrates ( Figure 1.3a). It has been proposed that the His458 and Asp206 residues can form hydrogen bonds with some of the substrates, helping to maintain them in the adequate conformation for electron transfer. Furthermore, computer simulations have shown that the H atom at ε-N in His458 is less than 5 Å away from the OH in phenolic substrates making it likely to participate in the electron transfer [20]. Once the T1 Cu 2+has been reduced to Cu 1+, it transfers electrons one by one to the cluster formed by the other three Cu atoms ( Figure 1.3b). This tri-nuclear cluster (TNC) is buried deeper inside the enzyme at the interface between two domains. In this cluster, oxygen is reduced to two molecules of water in a four-electron process that takes place as two sequential steps. First, the water is reduced in a two-electron process to a peroxide-level intermediate by the two T3 Cu 1+ions. Then, electrons from the T1 and T2 Cu 1+ions further reduce this intermediate to water ( Figure 1.3c) [21, 22].
Different sources for laccase include insects, bacteria, fungi and plants like the Japanese lacquer tree ( Toxicodendron vernicifluum ), where it was first extracted from [24] and which gives the enzyme its name. Laccase varieties extracted from fungi present the highest redox potentials [21, 22], and are thus preferred for biocathode development. Laccase isolated from different species presents some variations in their amino acid sequence [20]. Laccase from Trametes versicolor is particularly popular in electrochemistry research and thus, the numbering of the residues in the previous paragraph is referred to the numbering in this species.
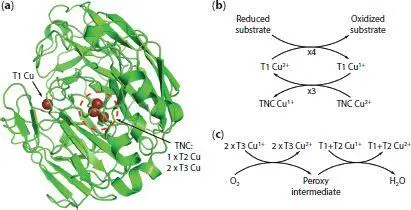
Figure 1.3 (a) Structure of laccase from Trametes versicolor elucidated by Bertrand et al . [23] (PDB code 1KYA), showing the surface T1 Cu and the buried trinuclear cluster (TNC). Mechanism of substrate oxidation (b) and oxygen reduction (c) by laccase.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Biofuel Cells»
Представляем Вашему вниманию похожие книги на «Biofuel Cells» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Biofuel Cells» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.