Georg Adler - Handbuch Demenzvorsorge
Здесь есть возможность читать онлайн «Georg Adler - Handbuch Demenzvorsorge» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на немецком языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Handbuch Demenzvorsorge
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Handbuch Demenzvorsorge: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Handbuch Demenzvorsorge»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Dieses Buch beschreibt die neurobiologischen Grundlagen der beginnenden Alzheimer-Demenz, ihre charakteristischen Frühsymptome und die Interpretation der Ergebnisse der psychologischen und technischen Untersuchungen. Maßnahmen zur körperlichen und geistigen Aktivierung und ein individuell angepasstes Risikofaktorenmanagement werden dargestellt. Die präventive Wirkung von Naturstoffen und Diäten wird differenziert bewertet.
Handbuch Demenzvorsorge — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Handbuch Demenzvorsorge», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Besorgniserregend ist in diesem Zusammenhang die zunehmende Häufigkeit von Übergewicht und Diabetes mellitus bei Jugendlichen und jungen Erwachsenen. Außer der individuellen Beratung und Behandlung der Betroffenen spielen die gesellschaftlichen Rahmenbedingungen für Ernährung und körperliche Aktivität eine große Rolle, wobei auch die Politik Einflussmöglichkeiten hat und Verantwortung trägt (Tataranni 2003).
Zusammenfassend lässt sich feststellen, dass die Identifikation beeinflussbarer Risikofaktoren für die Entwicklung einer Demenz und die Häufung dieser Risikofaktoren bei älteren Erwachsenen eine ausgezeichnete Basis für wirksame Maßnahmen zur Demenzprävention darstellt. Die daraus ableitbaren Maßnahmen betreffen sowohl den Lebensstil als auch die medizinische Behandlung der Risikofaktoren. Auf diese Weise könnte die durch den demographischen Wandel bedingte starke Zunahme von Demenzerkrankungen erheblich abgemildert werden.
2 Neurobiologie der Alzheimer-Demenz
Unter Demenz versteht man eine im Laufe des Lebens auftretende Störung des Kurzzeitgedächtnisses und der geistigen Leistungsfähigkeit, die zu einer Beeinträchtigung der für ein selbstständiges Leben erforderlichen Alltagsfertigkeiten führt. Ein derartiges Demenz-Syndrom kann verschiedene Ursachen haben, z. B. die Alzheimer-Krankheit, Durchblutungsstörungen des Gehirns oder andere neurodegenerative Erkrankungen. Aber auch allgemein körperliche Erkrankungen, wie z. B. eine ausgeprägte Anämie, oder andere Gehirnerkrankungen, wie z. B. ein Gehirntumor, können eine Demenz verursachen.

Abb. 2.1: Histopathologische Veränderungen bei der Alzheimer-Demenz (Alzheimer 1911). Zeichnungen Alzheimers, in der die Plaques und Fibrillen in der Großhirnrinde bei zwei Alzheimer-Patienten abgebildet sind.
Die bei weitem häufigste Ursache der Demenz ist jedoch die Alzheimer-Krankheit. Ihre Symptomatik und die zugrundeliegenden histopathologischen Veränderungen des Gehirns in Form von Plaques und Fibrillen wurden erstmalig 1906 von Alois Alzheimer beschrieben ( 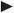 Abb. 2.1).
Abb. 2.1).
Mittlerweile ist bekannt, dass die Plaques überwiegend aus dem Eiweiß Beta-Amyloid, die Fibrillen überwiegend aus Tau-Protein bestehen. Die der Alzheimer-Krankheit zugrundeliegenden neurobiologischen Prozesse, die zur Ablagerung von Beta-Amyloid und Tau-Protein im Gehirn in Form von Plaques und Fibrillen führen, laufen über viele Jahre ab, bis sie schließlich zu Einschränkungen der geistigen Leistungsfähigkeit führen (Morris 2005).
2.1 Amyloid-Kaskaden-Hypothese
Die vorherrschende Theorie für die der Alzheimer-Demenz zugrundeliegenden neurobiologischen Prozesse ist die Amyloid-Kaskaden-Hypothese (Hardy und Higgins 1992). In ihrer ursprünglichen Form besagt sie, dass die Erkrankung durch die übermäßige Produktion, Aggregation und extrazelluläre Ablagerung von Beta-Amyloid, einem Abbauprodukt des Amyloid-Präkursor-Proteins (APP), ausgelöst wird. Als Folge der erhöhten Beta-Amyloid-Konzentration im Interstitium kommt es intrazellulär zur Bildung von Neurofibrillen aus hyperphosphoryliertem Tau-Protein. In den vergangenen Jahren ist die Amyloid-Kaskaden-Hypothese mit dem Zuwachs der Kenntnisse über die neurobiologischen Krankheitsabläufe weiterentwickelt worden (Hardy 2009).
Die überzeugendsten Hinweise für eine ursächliche Rolle von Beta-Amyloid in der Genese der Alzheimer-Krankheit stammen aus Studien bei den autosomal dominant vererbten familiären Alzheimer-Demenzen, die etwa 1–5 % der Krankheitsfälle ausmachen (Reitz und Mayeux 2014) und bei denen Mutationen der Gene für das Amyloid-Präkursor-Protein (APP), für Präsenilin 1 (PSEN1) oder für Präsenilin 2 (PSEN2) vorliegen (Guerreiro und Hardy 2014). Die bei diesen Alzheimer-Demenzen gewonnenen Erkenntnisse wurden auf die sogenannte sporadische Alzheimer-Demenz, die bei der ganz überwiegenden Anzahl der Patienten vorliegt, übertragen.
Das Amyloid-Präkursor-Protein (APP) ist ein großes Membranprotein, das insbesondere in den Synapsen von Nervenzellen vorkommt. Die physiologische Funktion des APP ist noch unbekannt; möglicherweise spielt es eine Rolle bei der synaptischen Plastizität. Bei der Verstoffwechselung von APP fällt Beta-Amyloid an, das durch Sekretasen aus dem APP herausgeschnitten wird. PSEN1 und PSEN2 sind katalytische Untereinheiten der Gamma-Sekretase, die an diesem Prozess beteiligt ist.
Wahrscheinlich besteht bei den familiären, früh beginnenden Formen der Alzheimer-Demenz eine genetisch bedingte Überproduktion von Beta-Amyloid, während bei den sporadischen, spät beginnenden Formen der Alzheimer-Demenz eher eine verminderte Elimination von Beta-Amyloid die treibende Kraft des Krankheitsprozesses ist (Mawuenyega et al. 2010). Die Elimination von Beta-Amyloid aus dem Gehirn findet einerseits durch Amyloid-abbauende Enzyme, andererseits durch verschiedene Transportmechanismen statt.
Die Mehrzahl der Amyloid-abbauenden Enzyme sind Zink-Metalloproteasen, unter anderem Neprilysin (NEP), Insulin-degrading enzyme (IDE) und Angiotensin-converting enzyme (ACE) (Nalivaeva und Turner 2019). Eine verminderte Aktivität des IDE, das im Gehirn sowohl Insulin als auch Beta-Amyloid hydrolysiert, ist vermutlich eine der Ursachen für die engen Zusammenhänge zwischen Alzheimer-Demenz und Typ-2-Diabetes mellitus (Steen et al. 2005).
Bei der Elimination von Beta-Amyloid aus dem Gehirn wirken auch Apolipoprotein E und Alpha-2-Makroglobulin mit, die den Transport von Beta-Amyloid durch die Blut-Hirn-Schranke in den Blutkreislauf fördern (Ries und Sastre 2016). Die Wirksamkeit der Elimination durch Apolipoprotein E ist dabei von der Isoform dieses Enzyms abhängig: die Isoform E2 ist wirksamer als E3, die wiederum wirksamer als E4 ist (Deane et al. 2008). Das Apolipoprotein E4 wirkt sich auf zweierlei Weise ungünstig auf das Fortschreiten der Alzheimer-Krankheit aus. Es ist zum einen mit einer verminderten Elimination von Beta-Amyloid aus dem Gehirn verbunden (Castellano et al. 2011), zum anderen zerfällt es zu neurotoxisch wirksamen Fragmenten (Mahley und Huang 2012).
Das Gehirn verfügt zur Gewebedrainage und zur Entfernung von Abfallprodukten an Stelle der Lymphgefäße über das sogenannte glymphatische System, ein perivaskuläres Netzwerk (Nedergaard 2013), das Anschluss zum lymphatischen System hat (Louveau et al. 2015). Lösliches Beta-Amyloid und Tau-Oligomere werden durch das glymphatische System aus dem Gehirn transportiert (Iliff et al. 2012, 2014). Die treibenden Kräfte im glymphatischen System sind Strömungs- und Diffusionsdruck (Benveniste et al. 2019). Die Elimination von Beta-Amyloid durch das glymphatische System ist im Tiefschlaf erhöht (Xie et al. 2013). Daher erhöht jegliche Art der Schlafstörung das Risiko für die Entwicklung einer Alzheimer-Demenz (Yaffe et al. 2014).
Die Ablagerungen von Beta-Amyloid im Gehirn folgen im Krankheitsverlauf einem charakteristischen raum-zeitlichen Muster (Nelson et al. 2009). Sie beginnen im basalen Neokortex und breiten sich später über den Hippokampus in den gesamten Kortex aus.
Eine erhöhte Konzentration von Beta-Amyloid im Interstitium wirkt neurotoxisch, führt zur Atrophie von Dendriten und Axonen und schließlich zum Zelluntergang (Yankner et al. 1990). Ein dabei wirksamer Mechanismus ist, dass Beta-Amyloid über den Caspase-Rezeptor die intrazelluläre Hyperphosphorylierung und Fibrillenbildung des Tau-Proteins fördert (Götz et al. 2001).
Die physiologische Funktion des Tau-Proteins besteht in der Stabilisierung der Mikrotubuli sowie in der Unterstützung des Wachstums der Neuriten und des intrazellulären Transports von Zellorganellen. Dabei wird seine Funktion physiologischerweise durch die von Kinasen und Phosphatasen katalysierte Phosphorylierung und Dephosphorylierung geregelt. Unter dem Einfluss von Beta-Amyloid wird das Tau-Protein hyperphosphoryliert. Dadurch kommt es zur Desintegration und zum Funktionsverlust der Neurotubuli, die die Straßen für den intrazellulären Transport von Zellorganellen darstellen. Dies führt zur Schädigung und schließlich zum Untergang der Nervenzellen (Reddy 2011). Das hyperphosphorylierte Tau-Protein wird unlöslich und aggregiert zu Filamenten und Fibrillen (Iqbal et al. 2005).
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Handbuch Demenzvorsorge»
Представляем Вашему вниманию похожие книги на «Handbuch Demenzvorsorge» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Handbuch Demenzvorsorge» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.