Heterogeneous Catalysts
Здесь есть возможность читать онлайн «Heterogeneous Catalysts» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Heterogeneous Catalysts
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Heterogeneous Catalysts: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Heterogeneous Catalysts»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
tate-of-the-art knowledge of heterogeneous catalysts including new applications in energy and environmental fields
This book focuses on emerging techniques in heterogeneous catalysis, from new methodology for catalysts design and synthesis, surface studies and operando spectroscopies, ab initio techniques, to critical catalytic systems as relevant to energy and the environment. It provides the vision of addressing the foreseeable knowledge gap unfilled by classical knowledge in the field.
Heterogeneous Catalysts: Advanced Design, Characterization and Applications
Presents recent developments in heterogeneous catalysis with emphasis on new fundamentals and emerging techniques Offers a comprehensive look at the important aspects of heterogeneous catalysis Provides an applications-oriented, bottoms-up approach to a high-interest subject that plays a vital role in industry and is widely applied in areas related to energy and environment
is an important book for catalytic chemists, materials scientists, surface chemists, physical chemists, inorganic chemists, chemical engineers, and other professionals working in the chemical industry.
Heterogeneous Catalysts — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Heterogeneous Catalysts», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Mean Au particle size, calculated by counting particles in high‐resolution TEM images of many different regions, is the crucial factor in determining activity. Results from recycled Au 55‐based catalysts confirm that deactivation does not occur. All reactions were carried out at 100 °C in toluene.
aCatalyst preparation: Au 55, Au 55preparation.
bExact loading was determined using inductively coupled plasma mass spectroscopy.
cFor full statistical breakdown of particle size distributions.
The importance of efficient activation of cluster‐based catalysts for CO oxidation was highlighted by a study that compared ligand removal using ozone exposure against a rapid thermal treatment for catalysts containing Au 13[PPh 3] 4[S(CH 2) 11CH 3] 4(8 Å diameter) ligand‐protected clusters on titania (anatase phase) [68]. The authors demonstrated that ozone treatment, followed by calcination at 400 °C for two hours in air, resulted in the most active catalyst, which had smaller gold particles compared with the less active catalyst treated by calcination alone.
The team of Professor Tsukuda used [Au 11(PPh 3) 8Cl 2]Cl, mentioned earlier [27], to prepare a series of mesoporous silica‐supported catalysts for oxidation of alcohols. The authors reported the importance of using a mixture of solvents (80% dichloromethane and 20% ethanol) in order to achieve homogeneous dispersion of clusters over a large surface area by optimizing the solvent‐mediated interaction of cluster with support [69]. Heat treatment at 200 °C produced active catalysts with Au clusters of ∼1 nm confined within a high surface area mesoporous silica support (SBA‐15) [69]. In another excellent study, the same team used hydroxyapatite (Ca 10(PO 4) 6(OH) 2) as a support for a range of thiol‐protected gold clusters Au n( n = 10, 18, 25, 39). The authors pinpointed a maximum of activity with an impressive turnover frequency (TOF) of 18 500 h −1per Au atom at n = 39 in solvent‐free peroxide‐initiated aerobic selective (∼99%) oxidation of cyclohexane to cyclohexanol and cyclohexanone [70]. The use of hydroxyapatite prevented cluster aggregation due to strong interaction between the Au clusters and PO 4 3−moieties. However, it proved impossible to differentiate catalysts with respect to cluster sizes since, in all samples, these appeared to be around 1 ± 0.5 nm in size even when sophisticated high‐angle annular dark field‐scanning TEM was used at moderate to high magnifications. This observation was explained by formation of “an ensemble of structural isomers produced in the calcination of Au n(SG) m,” which “will give rise to polydisperse TEM images.” Aberration‐corrected electron microscopy studies of pure Au 144supported on ultrathin carbon film, followed by reconstruction of obtained data, allowed construction of 3D electron density maps of such clusters [71]. Atomically resolved structures of Au 9clusters supported on TiO 2nanosheets ( Figure 5.6) obtained using aberration‐corrected electron microscopy were successfully matched with structural isomers of supported clusters predicted by high‐level quantum mechanical calculations [72].
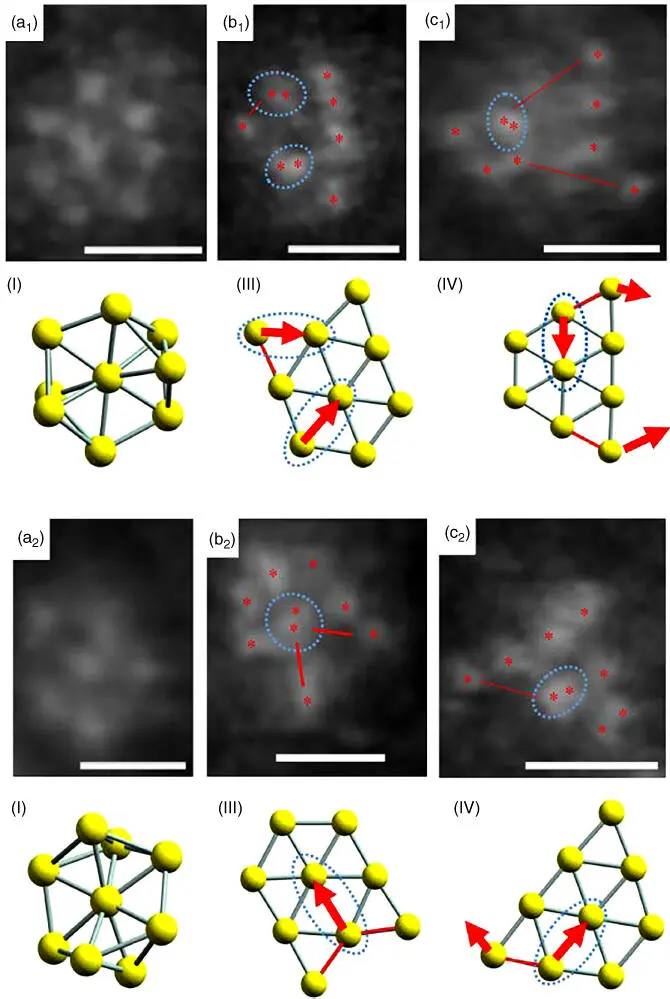
Figure 5.6 High‐resolution electron microscopy images (a1–c1) and (a2–c2) of Au 9clusters deposited on titania nanosheets under optimized imaging conditions. The scale bar is 0.5 nm. Panels I, III, and IV show the density functional theory (DFT) models for three isomers of Au 9.
Source: Al Qahtani et al. 2016 [72]. Reproduced with permission of American Institute of Physics. (See online version for color figure).
The smallest clusters are not always the most active species, where, in the case of benzyl alcohol oxidation, larger particles are more active: “unexpectedly, Au ∼144and Au ∼330on hierarchically porous carbon exhibited significantly higher turnover frequency than Au 25and Au 38” [73]. In fact, use of Au 9cluster‐based catalysts, which gradually evolved toward larger sizes, allowed us to establish a particle size effect in solvent‐free aerobic oxidation of cyclohexene – we showed that catalytic activity appeared only after Au 0particles larger than 2 nm had formed [74]. Such particle growth was observed only in the presence of trace amounts of peroxide species in the cyclohexene (stabilizer‐free reagent), highlighting the sensitivity of such catalysts in terms of particle size stability to the reaction media. This study also demonstrated that conventional bright‐field TEM is unsuited for the imaging of clusters on SiO 2NPs due to the poor contrast with the supported sub‐1 nm Au clusters. X‐ray photoelectron spectroscopy can be used to pinpoint the presence of clusters on supports since the characteristic electron binding energy is shifted to higher values in the case of ultrasmall clusters (regardless of their origin – made under UHV or chemically synthesized) [39].
Interestingly, even for the seemingly similar in nature Au n(SG) mclusters ( n = 10, 15, 18, 22, 25, 29, 33, 39), only some of the clusters ( n = 10, 15, 18, 25, 39) proved to be stable with respect to aggregation during activation, while others ( n = 22, 29, 33) produced larger aggregates under the same activation conditions [75]. Although catalysts with “as‐deposited” clusters had higher catalytic activity than the pure support, the removal of ligands resulted in about fourfold increase in the H 2production rate. Similarly, about fourfold higher activity of a Au 10‐cluster‐based catalyst was observed in comparison with catalysts made using large Au NPs.
In cases of relatively weak affinity between clusters and surface of support, fabrication of catalysts with reasonably high metal loadings may require a special approach of adding a non‐solubilizing (for cluster) solvent to the deposition mixture. For example, in the case of deposition of the Au 9on WO 3for loadings greater than 0.1 wt%, n ‐hexane was slowly added to the mixture of gold cluster and WO 3in CH 2Cl 2to ensure cluster deposition [76].
A final contemporary example is the use of cleverly designed Au 25‐loaded BaLa 4Ti 4O 15water splitting photocatalyst with the cluster‐based active sites protected by chromium oxide shell for enhanced activity and stability [77]. The chromium oxide shell is impermeable to O 2but permeable to H +, thus allowing the distinction of active sites for the evolutions of H 2(by photoelectron reduction) and O 2(by photohole oxidation). This resulted in a 19‐fold improvement in performance and excellent longevity of the catalyst due to the prevention of gold cluster sintering.
Mixed metal clusters offer the advantages of synergistic action and cooperativity by two different metals [53]; a recent highly comprehensive review covers the advances in the use of heterometallic cluster‐based catalysts [78]. As an example to illustrate that “each atom counts,” a series of chemically synthesized metal cluster catalysts were made using a family of clusters differing by an atom or two from each other – Ru 6Sn, Ru 5Pt, Ru 5PtGe, and Ru 5PtSn [36]. The obtained materials were tested ( Figure 5.7) for the single‐step conversion of dimethyl terephthalate (DMT) into cyclohexanedimethanol (CHDM). The Ru 5PtSn‐based catalyst demonstrated superior activity, as evidenced by the highest conversion of the tested catalysts, as well as excellent selectivity toward the desired product CHDM. The only substantial by‐product is dimethyl hexahydroterephthalate (DMHT), which is an intermediate of the hydrogenation pathway. Further optimization of the catalyst design and catalytic test conditions could result in even better performance. For a detailed and critical update on specific examples of a wide range of cluster‐based catalysts and their performance in various catalytic processes, curious readers are referred to a monumental recent review by Liu and Corma [79].
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Heterogeneous Catalysts»
Представляем Вашему вниманию похожие книги на «Heterogeneous Catalysts» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.

Обсуждение, отзывы о книге «Heterogeneous Catalysts» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.