Recent Advances in Polyphenol Research
Здесь есть возможность читать онлайн «Recent Advances in Polyphenol Research» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Recent Advances in Polyphenol Research
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Recent Advances in Polyphenol Research: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Recent Advances in Polyphenol Research»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Recent Advances in Polyphenol Research
Recent Advances in Polyphenol Research — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Recent Advances in Polyphenol Research», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
An asymmetric version of this approach appeared recently by using a salt of binaphthol‐derived chiral phosphoric acid as a chiral phase transfer catalyst ( Figure 2.17) (Yang et al. 2016). Reaction of flavylium salt 28with phloroglucinol derivative 29in the presence of Aas a catalyst (10 mol%) allowed the enantioselective nucleophilic attack of 29to the C(4) position of 28to give adduct 30, which was treated with p ‐toluenesulfonic acid to give bicycle 31in 56% yield with a high enantioselectivity (94% ee ).
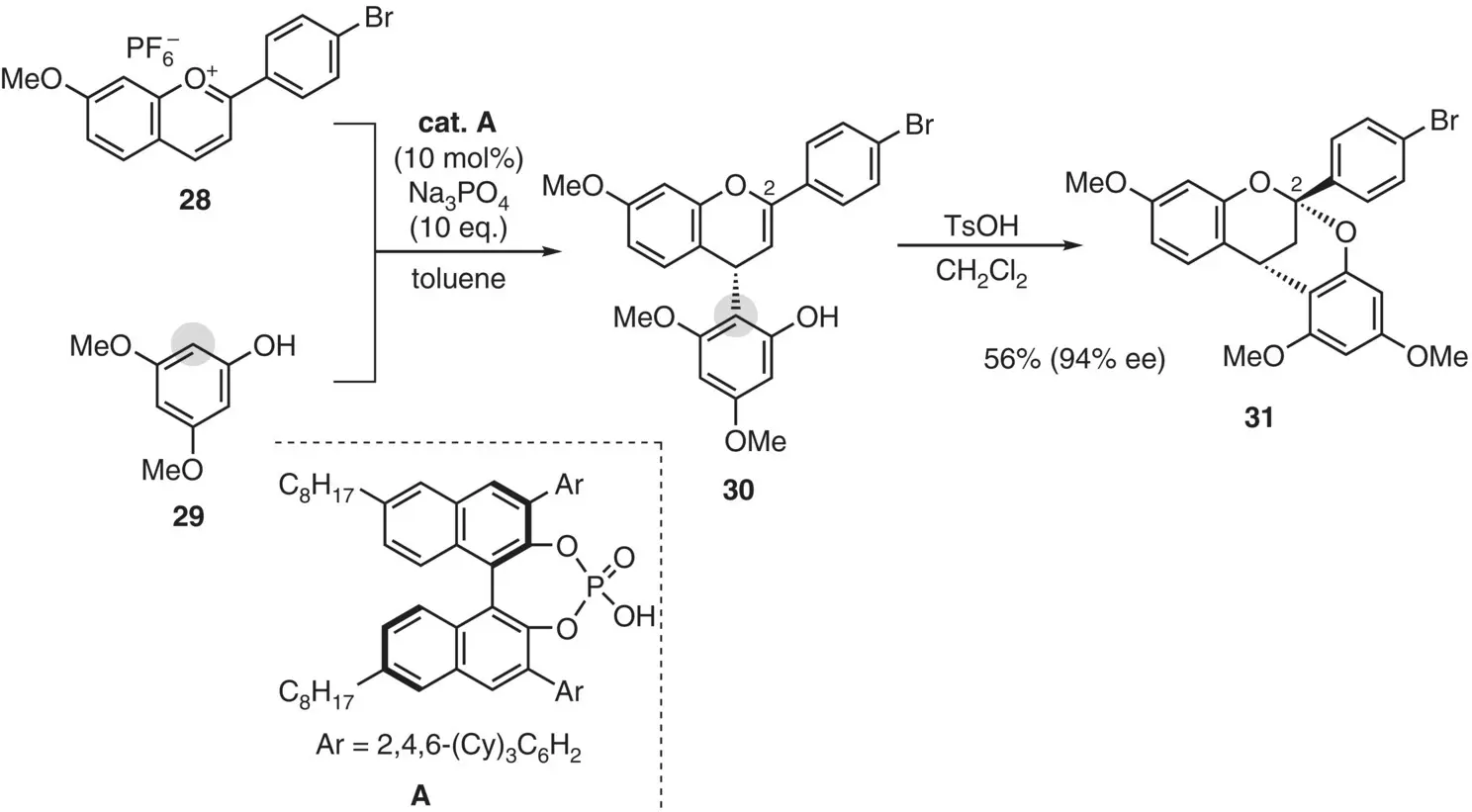
Figure 2.17 Asymmetric annulation approach.
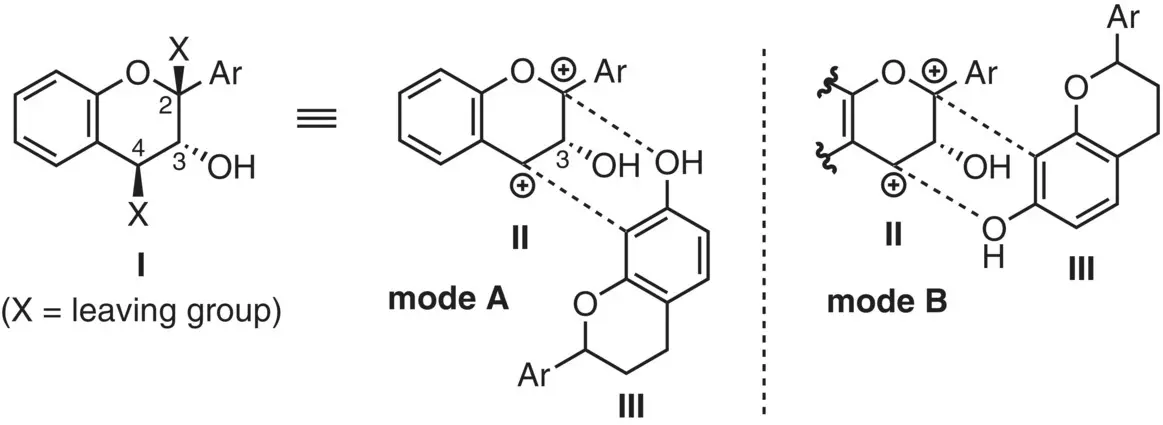
Figure 2.18 Strategy for stereoselective annulation.
The present authors' group recently developed a novel concept in a flavan annulation ( Figure 2.18) (Ito et al. 2014). Two requirements en route to the requisite dioxabicyclic skeleton include (i) design of a suitable precursor Ito generate the dicationic species II, and (ii) regioselective dual bond formation with IIIvia mode A, not vice versa (mode B). An additional requirement was the stereoselectivity. Thanks to the C(3) stereocenter in II, the annulation would proceed in a stereoselective manner.
As a dication equivalent, two oxygen‐based leaving groups X at the C(2) and C(4) positions were envisioned, and the precursor Icould be available by oxidation of flavan derivatives. Thus, the oxidation of epicatechin derivative 32with DDQ in the presence of ethylene glycol gave 2,4‐ethylenedioxy derivative 33in 69% yield ( Figure 2.19). The C(8) position of 33was masked by a bromine atom, affording bromide 34in excellent yield.
As a model study using the dication precursor 34,the reaction with phloroglucinol derivative 35was examined in the presence of BF 3·OEt 2( Figure 2.20). Upon the reaction started at –78 °C followed by gradual warming, the starting material 34was consumed at –40 °C, and two products were produced (run 1, Table 2.1). The major product was single‐linked compound 36with the C(4)–C(2) bond, whereas the minor product 37had the desired dioxabicyclic structure. Importantly, these products, 36and 37, were the single stereoisomers, respectively. Re‐exposure of 36to the same conditions led to a smooth conversion into 37. Thus, the annulation proceeded in a stepwise manner, starting with the formation of the C(4)–C(2) bond from the β‐side to form 36followed by the C(2)–O bond formation to give 37. Indeed, by extending the reaction time and raising the temperature, the annulation of 34and 35went to completion, giving 37in excellent yield (run two).
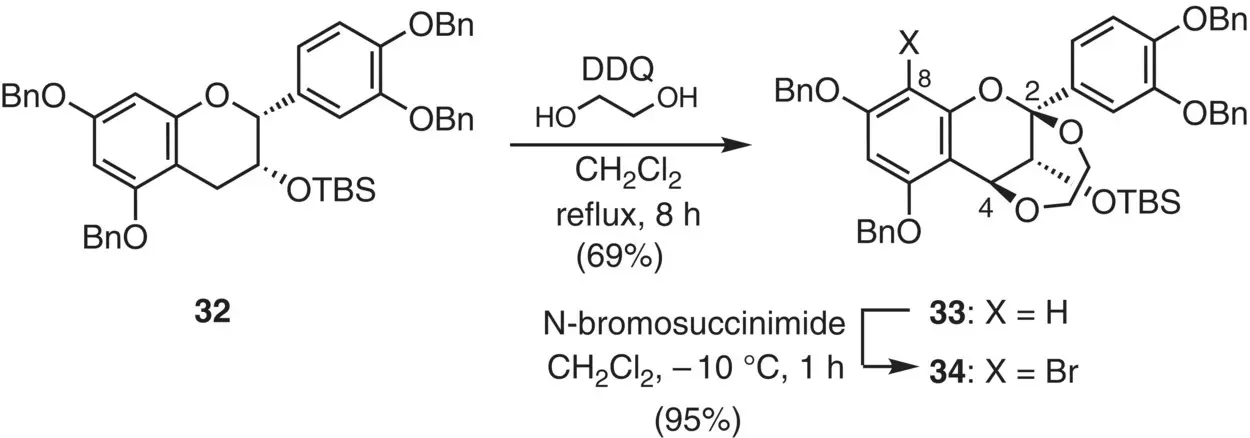
Figure 2.19 Synthesis of a 2,4‐dioxy flavan derivative.

Figure 2.20 Model study for stereoselective flavan annulation.
Table 2.1 Results of flavan annulation.
| run | time / h a) | temperature / °C | product (yield) |
|---|---|---|---|
| 1 | 2 | −78 → −40 | 36(66%), 37(20%) |
| 2 | 3 | −78 → −20 | 37(93%) |
a)Duration of the gradual warming.
2.3.6 Total Synthesis
This section describes several completed syntheses of the oligomeric PAs with A‐type linkages.
2.3.6.1 Synthesis of Diinsininol Aglycon (45)
Selenski and Pettus (2006) developed an efficient synthetic approach to bicycle 45, corresponding to the aglycon of diinsininol ( 46), by exploiting the [3+3] annulation approach ( Figure 2.21). As an electrophilic unit, flavylium 40was prepared by the condensation of 2,4,6‐triacetoxybenzaldehyde ( 38) and 3,4‐dihydroxyacetophenone ( 39). On the other hand, the nucleophilic partner 44was synthesized from styrene 41and benzaldehyde 42. Treatment of 42with LiAlH 4in the presence of MgBr 2generated o ‐quinonemethide A, which underwent a [4+2]‐cycloaddition with styrene 41, giving the corresponding flavan 43in 45% yield. DDQ‐oxidation in the presence of H 2O followed by removal of the protecting groups afforded free flavanone 44in 72% yield (two steps). The reaction of flavylium salt 40with nucleophile 44(6 equiv) proceeded under microwave irradiation at 120 °C, giving the annulated product 45in 32% yield. The product was obtained as a single diastereomer, albeit in a racemic form.
2.3.6.2 Synthesis of Procyanidin A2 (3)
The protocol described earlier ( Figure 2.20) was applied to the synthesis of procyanidin A2 ( 3) (see Figure 2.5), in which a nucleophilic monomer unit needed to be selectively protected ( vide infra ). The present authors have developed a de novo synthetic approach to the epi‐type catechins ( Figure 2.22) (Stadlbauer et al. 2012), relying on the ortho ‐metalation of aryl fluoride I(#1) and reaction with epoxy alcohol II, followed by an S NAr oxycyclization of adduct IIIto give flavan IV(#2). This protocol enabled access to monomer unit 52with a free hydroxy group at C(7) ( Figure 2.23) (Ito et al. 2014). Starting from 1,3,5‐trifluorobenzene ( 47), sequential substitutions with t ‐BuOK and BnOK gave mono‐fluoride 48. Regioselective lithiation of 48followed by the reaction with epoxy alcohol 49in the presence of BF 3·OEt 2gave adduct 50. After the protecting group arrangement, the resulting alcohol 51was subjected to the pyran‐ring formation. Treatment of 51with KH cleanly afforded the cyclized product, which was exposed to acidic conditions to give the monomer unit 52, which was employed as the nucleophilic flavan unit in the following annulation ( Figure 2.24). Upon treatment of a mixture of 34and 52with BF 3·OEt 2, the corresponding annulation product 53was obtained in excellent yield. Notably, none of other regio‐ and stereoisomers was observed. As the α‐face of the electrophilic flavan unit 34is shielded by the C(3)‐substituent, nucleophile 52approached from the β‐side to form the double linkages at the C(2) and C(4) positions. In addition, the nucleophile 52selectively reacted at its C(8) position rather than the C(6) position. Finally, all protecting groups were removed to give procyanidin A2 ( 3).
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Recent Advances in Polyphenol Research»
Представляем Вашему вниманию похожие книги на «Recent Advances in Polyphenol Research» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Recent Advances in Polyphenol Research» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.