David Machin - Randomised Clinical Trials
Здесь есть возможность читать онлайн «David Machin - Randomised Clinical Trials» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Randomised Clinical Trials
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Randomised Clinical Trials: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Randomised Clinical Trials»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
sed Clinical Trials: Design, Practice and Reporting
This second edition contains extensively revised material throughout, including new chapters covering designs for repeated measures, non-inferiority, cluster and stepped wedge trials. Other new chapters describe data and safety monitoring, biomarker studies, and feasibility studies. Updated and expanded sections discuss situations where multiple organs, different body locations or competing risks are involved, subgroup analysis, and multiple outcomes. Written by an author team with extensive experience in conducting clinical trials, this book:
Provides comprehensive coverage of randomised clinical trials, ranging from basic to advanced Features several new chapters, updated case studies and examples, and references to changes in regulations Explains basic randomised trials, including the parallel two-group controlled trial with a single outcome measure Covers paired trial designs and trials with more than two interventions Includes a chapter on miscellaneous topics such as adaptive designs, large simple trials, Bayesian methods for very small trials, alpha-spending functions and the predictive probability test is essential reading for clinicians, nurses, data managers, and medical statisticians involved in clinical trials, and for health practitioners responsible for direct patient care in a clinical trial setting.
Randomised Clinical Trials — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Randomised Clinical Trials», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
The proper use of randomization guarantees that there is no bias in the selection of patients for the different treatments and so helps considerably to reduce the risk of differences in experimental environment. Randomized allocation is not difficult to implement and enables trial conclusions to be more believable than other forms of treatment allocation.
As a consequence, we are focussing on randomised controlled trials and not giving much attention to less scientifically rigorous options.
1.3.3 Design hierarchy
The final choice of design for a clinical trial will depend on many factors, key amongst these are clearly the specific research question posed, the practicality of recruiting patients to such a design and the resources necessary to support the trial conduct. We shall discuss these and other issues pertinent to the design choice in later chapters. Nevertheless, we can catalogue the main types of design options available and these are listed in Figure 1.2. This gives a relative weight to the evidence obtained from these different types of clinical trial. All other things being equal, the design that maximises the weight of the resulting evidence should be chosen. For expository purposes, we assume that a comparison of a new test treatment with the current standard for the specific condition in question is being made.
1.3.3.1 Randomisation
The design that provides the strongest type of evidence is the double‐blind ( or double‐masked ) randomised controlled trial (RCT). In this, the patients are allocated to treatment at random and this ensures that in the long run patients, before treatment commences, will be comparable in the test and standard groups. Clearly, if the important prognostic factors that influence outcome were known, one could match the patients in the standard and test groups in some way. However, the advantage of randomisation is that it balances for unknown and the known prognostic factors and this could not be achieved by matching. Thus, the reason for the attraction of the randomised trial is that it is the only design that can give an absolute certainty that there is no bias in favour of one group compared to another at the start of the trial. Indeed, in Example 1.12, Erbel, Di Mario, Bartunek, et al . (2007), who essentially conducted a single‐arm prospective case study, admitted that failure to conduct a randomised comparison compromised their ability to draw definitive conclusions concerning the stent on test.
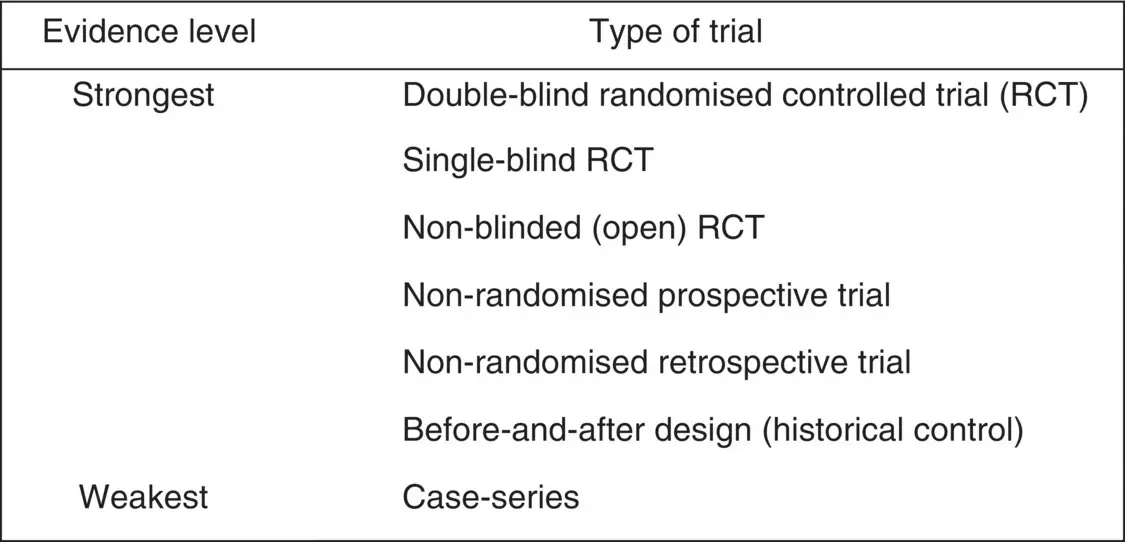
Figure 1.2The relative strength of evidence obtained from alternative designs for comparative clinical trials
1.3.3.2 Blinding or masking
For the simple situation in which the attending clinician is also the assessor of the outcome, the trial should ideally be double‐blind (or double‐masked). This means that neither the patient nor the attending clinician will know the actual treatment allocated. Having no knowledge of which treatment has been taken, neither the patient nor the clinician can be influenced at the assessment stage by such knowledge. In this way, an unprejudiced evaluation of the patient response is obtained. Thus Meggitt, Gray and Reynolds (2006) used double‐blind formulations of Azathioprine or Placebo so that neither the patients with moderate‐to‐severe eczema, nor their attending clinical team, were aware of who received which treatment. Although they did not give details, the blinding is best broken only at the analysis stage once all the data had been collated.
Despite the inherent advantage of this double‐blind design, most clinical trials cannot be conducted in this way as, for example, a means has to be found for delivering the treatment options in an identical way. This may be a possibility if the standard and test are available in tablet form of identical colour, shape, texture, smell and taste. If such ‘identity’ cannot be achieved, then a single‐blind design may ensue. In such a design, one of the patient or the clinical assessor has knowledge of the treatment being given but the other does not. In trials with patient survival time as the endpoint, double‐blind usually means that both the patient and the treating physician and other staff are blinded. However, assessment is objective (death) and the blinding irrelevant by this stage.
Finally, and this is possibly the majority situation, there will circumstances in which neither the patient nor the assessor can be blind to the treatments actually received. Such designs are referred to as ‘open’ or ‘open‐label’ trials.
1.3.3.3 Non‐randomised designs
In certain circumstances, when a new treatment has been proposed for evaluation, all patients are recruited prospectively but allocation to treatment is not made at random. In such cases, the comparisons may well be biased and hence are unreliable. The bias arises because the clinical team choose which patients receive which intervention and in so doing may favour (even subconsciously) giving one treatment to certain patient types and not to others. In addition, the requirement that all patients should be suitable for all options may not be fulfilled – in that if it is known that a certain option is to be given to a particular subject then one may not so rigorously check if the other options are equally appropriate. Similar problems arise if investigators have recruited patients into a single‐arm study, and the results from these patients are then compared with information on similar patients having (usually in the past) received a relevant standard therapy for the condition in question. However, such historical comparisons are likely to be biased also and to an unknown extent so again it will not be reasonable to ascribe the difference (if any) observed entirely to the treatments themselves. Of course, in either case, there will be situations when one of these designs is the only option available. In such cases, a detailed justification for not using the ‘gold standard’ of the randomised controlled trial is required.
Understandably, in this era of EBM, information from non‐randomised comparative studies is categorised as providing weaker evidence than that from randomised trials.
The before‐and‐after design is one in which, for example, patients are treated with the Standard option for a specified period and then, at some fixed point in time, subsequent patients receive the Test treatment. This is the type of design used by Erbel, Di Mario, Bartunek, et al . (2007) to evaluate a bioabsorbable stent for coronary scaffolding. In such examples, the information for the Standard is retrospective in nature and is often obtained from clinical records only and so was not initially collected for trial purposes. If this is the case, the before‐and‐after design is likely to be further compromised as, for example, in the ‘before’ period, the patient selection criterion, clinical assessments and data recorded may not meet the standards required of the ‘after’ component. Such differences are likely to influence the before‐and‐after comparison in unforeseen and unknown ways.
Example 1.14 Glioblastoma in the elderly – non‐randomised design
Brandes, Vastola, Basso, et al . (2003) describe a study comparing radiotherapy alone (Group A), radiotherapy and the combination of procarbazine, lomustine and vincristine (Group B) and radiotherapy with temozolomide (Group C) in 79 elderly patients with glioblastoma. The authors’ state:
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Randomised Clinical Trials»
Представляем Вашему вниманию похожие книги на «Randomised Clinical Trials» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.











Обсуждение, отзывы о книге «Randomised Clinical Trials» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.