Genetic Analysis of Complex Disease
Здесь есть возможность читать онлайн «Genetic Analysis of Complex Disease» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Genetic Analysis of Complex Disease
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Genetic Analysis of Complex Disease: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Genetic Analysis of Complex Disease»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
An up-to-date and complete treatment of the strategies, designs and analysis methods for studying complex genetic disease in human beings Genetic Analysis of Complex Diseases
Genetic Analysis of Complex Diseases
Genetic Analysis of Complex Diseases
Genetic Analysis of Complex Disease — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Genetic Analysis of Complex Disease», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Duchenne and Becker Muscular Dystrophy
Duchenne muscular dystrophy (DMD) is a severe, childhood‐onset X‐linked muscular dystrophy. Becker muscular dystrophy (BMD) is allelic with DMD but typically has a later age of onset and a milder presentation. Boys with DMD typically have normal development for the first few years of life, after which rapidly progressive muscle deterioration becomes obvious. Affected males usually lose the ability to walk by age 10–12 years. The eventual loss of muscle strength in the cardiac and respiratory muscles leads to death in early adulthood. Duchenne and Becker muscular dystrophy is caused by mutations in the DMD gene, which codes for the protein dystrophin (Koenig et al. 1988). Large deletions in this gene account for approximately 60–70% of cases of DMD and BMD; however, duplications and point mutations have also been reported (Takeshima et al. 2010). Mutations in this gene that alter the reading frame typically cause DMD, while mutations that preserve the reading frame lead to BMD. Researchers are investigating a variety of therapies including exon‐skipping and read‐through of stop codons.
Cystic Fibrosis
Cystic fibrosis (CF), an autosomal recessive disorder, is the most common hereditary disease among Northern Europeans, with a carrier frequency of between 1/25 and 1/30 (Hamosh et al. 1998). This condition affects the function of the pancreas, lungs, and sweat glands, among other organ systems (Ratjen and Doring 2003). In Northern Europeans, a single mutation in the CFTR gene called ΔF508 accounts for about 70% of the abnormal CF alleles. Three bp (codon 508) are deleted, and the resulting amino acid sequence is missing a phenylalanine. Although the reading frame is preserved in this particular mutation, the deletion results in a block in protein processing. CF is an excellent example of allelic heterogeneity in which different mutations, or alleles, at the same locus can cause a disease. In fact, more than 1900 other deleterious mutations in CFTR have been identified (Cystic Fibrosis Mutation Database 2015).
Charcot‐Marie‐Tooth Disease
Charcot‐Marie‐Tooth (CMT) Type 1A and hereditary liability to pressure palsies (HNPP) are caused by an abnormal 1.4 megabase CNV on 17p12. CMT is a peripheral neuropathy most commonly caused by a duplication of PMP22, while a deletion of this same gene will result in the phenotypically distinct condition, HNPP. There are numerous types of CMT, many of which are caused by mutations at many different loci, a scenario described as locus heterogeneity.
Nucleotide Repeat Disorders
Certain loci in the genome have variable numbers of nucleotide repeats, typically of di‐ or tri‐nucleotide length. Most are not associated with expressed genes but can be exploited as genetic markers since they are highly polymorphic. A few loci with tri‐nucleotide repeats are located near or within a gene and, by expansion beyond a certain threshold, can disrupt gene expression and cause disease.
More than 20 disorders have been shown to be the result of unstable nucleotide repeats (La Spada & Taylor 2010; Polak et al. 2013). The most common of these conditions are summarized in Table 2.4. Expansions may occur in the exon, intron, or regulatory elements of a gene. The phenotypes of some of these conditions, including myotonic dystrophy, Huntington disease, and Machado–Joseph disease, among others, are associated with anticipation, a clinical phenomenon in which disease severity worsens in each successive generation. Because disease expression can be quite variable and difficult to measure, age of onset is frequently utilized as an analogue of severity and anticipation is then observed as a decreasing mean age of onset with each passing generation. The anticipation seen in these disorders is attributed to the intergenerational instability and resulting expansion of the repeated elements. However, some repeat disorders are stable during mitosis and meiosis and therefore do not exhibit anticipation. Furthermore, certain conditions are more likely to exhibit an expansion only when transmitted from a particular sex. For instance, the CAG repeat in Huntington disease typically only expands in male gametes, while the CTG repeats in Fragile X usually only expands in female gametes.
Susceptibility Versus Causative Genes
As the study of common and genetically complex human diseases identifies the significant contribution of heredity in their development, it is likely that more genes or genetic risk factors will be found to affect susceptibility to disease rather than the more traditionally considered causative genes. Historical successes in the localization of genes have been with diseases whose mode of inheritance is known (as illustrated above). These disorders are often highly or completely penetrant and are due to a defect in a single gene, yet these Mendelian disorders are often relatively rare in the population. However, in recent years, genomic research has uncovered genetic risk factors for many diseases that were suspicious for genetic etiology but were unexplained by traditional Mendelian cause and effect. Some of the most common and deadly diseases of society, such as cardiovascular disease, cancer, and obesity, have significant genetic components that are evident from non‐random family clustering. These diseases are termed “complex” because they are likely due to the interaction of multiple factors, both environmental and genetic. Susceptibility genes for such complex disorders are substantially harder to identify than genes responsible for Mendelian disorders.
A well‐characterized example of a susceptibility locus is that of the apolipoprotein E (APOE) gene and Alzheimer disease (AD). The APOE gene on chromosome 19 has three different alleles, scored as 2, 3, and 4, which occur with frequencies 6%, 78%, and 16% in most European populations, respectively (e.g. Saunders et al. 1993). These alleles differ in their DNA sequence by only one base at codons 112 and/or 158 ( Figure 2.11). The APOE 4 allele increases risk and decreases age of onset in familial and sporadic late‐onset AD and early‐onset sporadic AD. The 2 allele has been shown to be protective to some extent for risk to develop AD (Corder et al. 1994, 1995a,b; Farrer et al. 1997). It is important to note that for APOE and AD, the 4 allele is not by itself sufficient or necessary for the development of AD but has been shown to be associated with increased susceptibility to AD.
Table 2.4 Salient features of human repeat expansion diseases.
| Condition | Gene symbol | Repeat type | Repeat localization | Repeat number abnormal range | Inheritance pattern | Clinical features |
|---|---|---|---|---|---|---|
| Fragile X syndrome | FRAXA | CGG | 5’ Untranslated region | 200–1000 (premutation range of 52–200) | X‐linked | Moderate to severe mental retardation, behavioral abnormalities, macroorchidism, large ears, and prominent jaw |
| Huntington disease (HD) | HD | CAG | Open reading frame | >36 (premutation range 27–35) | Autosomal dominant; expansion more common in paternal allele | Choreiform movements, dystonia, psychiatric illness, cognitive decline, dementia |
| Myotonic dystrophy | DM1 | CTG | 3’ Untranslated region | Mild: 50–150 Classic: ~100–1000 Congenital: >2000 | Autosomal dominant; expansion more common in maternal allele | Weakness, myotonia, ptosis, cataracts, cardiac arrhythmia; endocrine abnormalities, frontal balding |
| Friedrich Ataxia | FXN | GAA | Intron | 66–1700 uninterrupted repeats (premutation range of 34–65 uninterrupted repeats) | Autosomal recessive | Ataxia, sensory loss, weakness, diabetes mellitus, cardiomyopathy |
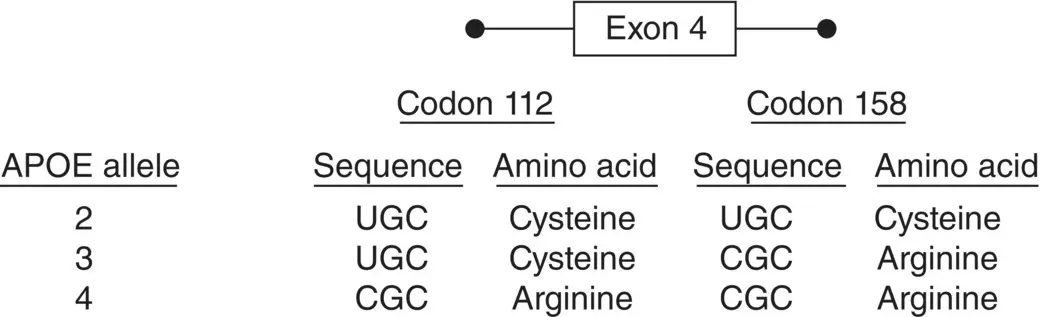
Figure 2.11 Single base pair changes in exon 4 of APOE define the 2, 3, and 4 alleles at this locus.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Genetic Analysis of Complex Disease»
Представляем Вашему вниманию похожие книги на «Genetic Analysis of Complex Disease» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.











Обсуждение, отзывы о книге «Genetic Analysis of Complex Disease» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.