Contemporary Accounts in Drug Discovery and Development
Здесь есть возможность читать онлайн «Contemporary Accounts in Drug Discovery and Development» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Contemporary Accounts in Drug Discovery and Development
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Contemporary Accounts in Drug Discovery and Development: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Contemporary Accounts in Drug Discovery and Development»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
A useful guide for medicinal chemists and pharmaceutical scientists Contemporary Accounts in Drug Discovery and Development
Contemporary Accounts in Drug Discovery and Development
Contemporary Accounts in Drug Discovery and Development
Contemporary Accounts in Drug Discovery and Development — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Contemporary Accounts in Drug Discovery and Development», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
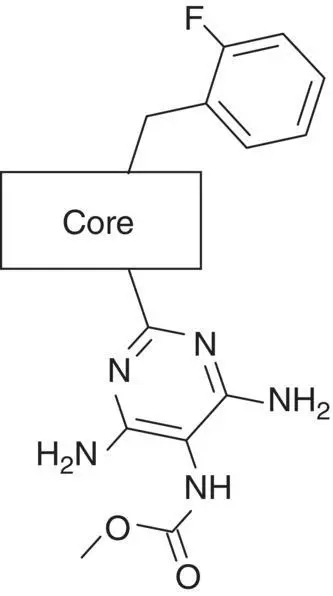
Table 3.4 Properties of the core variation compounds 16– 20, 2.
 |
||||
|---|---|---|---|---|
| Compound | Core | cGMP formation MEC a(μM) | In vitro clearance (rat hepatocytes) CL b(l/h/kg) | In vivo clearance (rat) CL b(l/h/kg) |
| 16 | 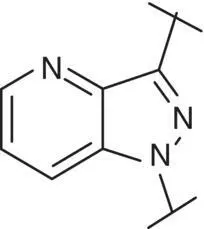 |
1.2 | <0.1 | 1.0 |
| 17 | 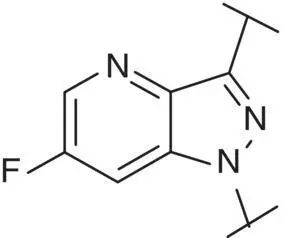 |
0.5 | 0.1 | 0.3 |
| 18 | 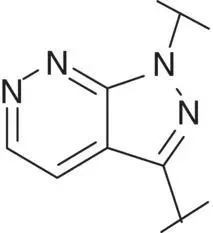 |
2.0 | <0.1 | 3.8 |
| 19 | 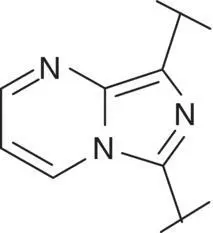 |
1.7 | n.d. b | 1.8 |
| 20 | 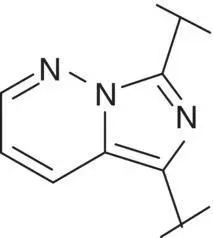 |
0.7 | <0.1 | 0.9 |
| 2 | 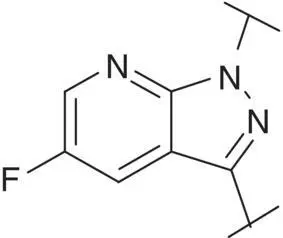 |
0.3 | 0.1 | 0.3 |
aMEC, minimal effective concentration to achieve stimulation of cGMP formation (≥3‐fold increase in basal luminescence) in a recombinant sGC‐overexpressing cell line [36].
bn.d., not determined.
Novel 1 H ‐pyrazolo[3,4‐ c ]pyridazine 18proved to be a reasonably potent sGC stimulator and seemed to be stable in rat hepatocytes (although solubility was limited and that might have impacted the assay) but exhibited an unacceptable high clearance of 3.8 l/h/kg when tested in vivo in rats and thus was not further profiled, along with imidazo[1,5‐ a ]pyrimidine 19for similar reasons. In contrast, imidazo[1,5‐ b ]pyridazine 20exhibited potent sGC stimulator properties (MEC = 0.7 μM), good metabolic stability in rat hepatocytes and a low to moderate clearance of 0.9 l/h/kg after i.v. dosing to rats. Finally, revisiting 1 H ‐pyrazolo[3,4‐ b ]pyridine 10with an additional fluorine at the 5‐position resulted in the potent sGC stimulator 2(vericiguat) (MEC = 0.3 μM) with good metabolic stability in rat hepatocytes and a surprisingly low clearance of 0.3 l/h/kg after i.v. dosing to rats ( Table 3.4). Thus, the blood clearance of derivative 10in rats (1.2 l/h/kg) was reduced fourfold by fluorination at position 5.
Finally, compounds 17, 20and 2were selected for further pharmacokinetic profiling across different species ( Table 3.5). Fluoropyrazolo[3,4‐ b ]pyridine derivative 2exhibited the best overall pharmacokinetic profile by far [40], with a low clearance and long half‐life in rats and dogs after i.v. dosing, as well as high oral bioavailability. In addition, 2had no inhibitory effects on major CYP isoforms (1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4), as indicated by IC 50values of >50 μM. Metabolite identification in human hepatocytes was characterized by a low turnover, with the glucuronide (major) and the debenzylated compound (minor) being the only metabolites found. Thus, the main biotransformation pathway shifted from predominantly phase 1 oxidative CYP‐mediated metabolism ( 1) to primarily phase 2 UGT‐mediated conjugation with glucuronic acid.
3.4 Synthesis Routes toward Vericiguat
3.4.1 Medicinal Chemistry Route to Vericiguat
The first vericiguat synthesis stemed from a time when rapid and flexible access to many derivatives was key in the project. For this purpose we employed the pyrazolo[3,4‐ b ]pyridine‐3‐iodide 21and coupled with 2‐chloro‐5‐nitropyrimidine‐4,6‐diamine, 34, by means of a modified‐Stille coupling using excess amounts of hexabutylditin and 32 mol% of palladium catalyst. Although yielding the desired product 23in low yields and moderate purity, it was very clear already at that time that this route will not be suitable for scale‐up campaigns for obvious reasons. Nevertheless, after reduction of the respective nitro group to the amine 24, a chemoselective acylation could be performed leading to 2in overall good yield for the last two steps ( Scheme 3.1).
Table 3.5 In vivo pharmacokinetic properties of 17, 20, vericiguat 2in comparison with riociguat 1.
| Compound | Species | V ss(l/kg) | CL b(l/h/kg) | t 1/2(h) | Bioavailability (%) |
|---|---|---|---|---|---|
| 17 | Rat | 0.5 | 0.3 | 1.5 | 26 |
| Dog | 1.0 | 0.2 | 4.1 | 56 | |
| 20 | Rat | 0.3 | 0.9 | 0.5 | 37 |
| Dog | 2.0 | 0.9 (Cl p) a | 1.8 | n.d. b | |
| 2 | Rat | 1.0 | 0.3 | 3.4 | 65 |
| Dog | 1.4 | 0.2 | 6.2 | 75 | |
| 1 | Rat | 1.2 | 1.3 | 1.4 | 46 |
| Dog | 0.7 | 0.3 (Cl p) a | 2.4 | 79 |
aCl p, plasma clearance.
bn.d., not determined.
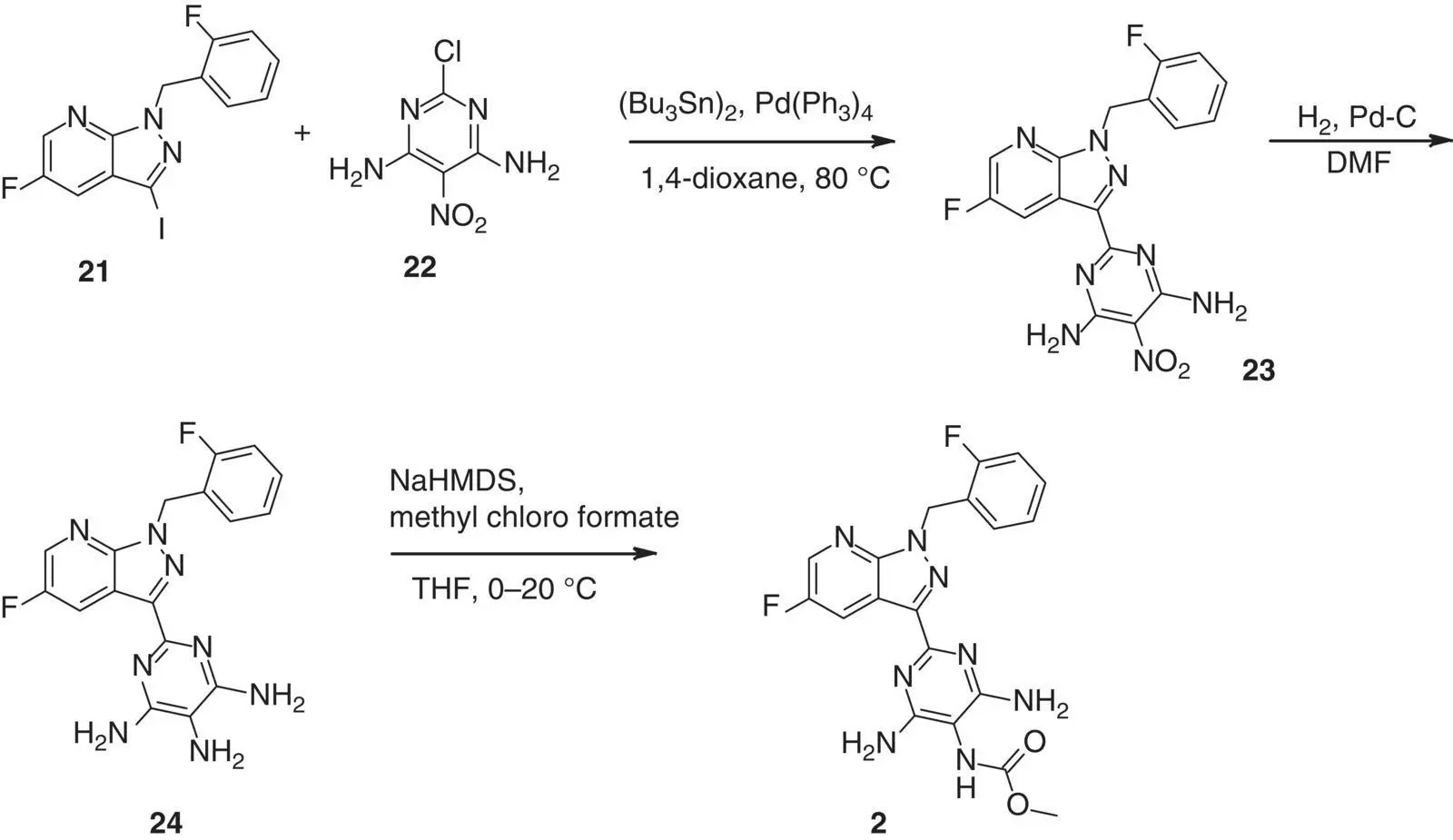
Scheme 3.1 Early days synthesis of vericiguat 2.
In the further course of the project, we decided to abandon the Stille coupling, which appeared rather unpredictable in terms of yields depending on the scale and thought of a more sustainable and scalable synthetic route with more reliable yields. Thus, the iodide 21was treated with copper(I) cyanide at 150 °C in DMF to yield the cyano derivative 25. In order to facilitate the formation of the pyrimidine, the cyano functional group was converted into an amidine using standard methodology. Based on experience from the former riociguat project it was planned to transform the amidine 26to the trisamino pyrimidine derivative 24using a reaction sequence highlighting a rarely used malonodinitrile derivative 27for condensation with the amidine 26and subsequent reduction to the desired trisamino derivative 24. We were pleased to find that this sequence worked nicely also for the novel fluoro core and it proved to be a more efficient route from iodide 21in our hands ( Scheme 3.2).
The synthesis leading to iodide 21started with a chemoselective dehalogenation of 2,6‐dichloro‐5‐fluoro‐3‐cyanopyridine 29which can be achieved by the transformations shown in Scheme 3.3. The 5‐fluoro‐1 H ‐pyrazolo[3,4‐ b ]pyridine 33was constructed by reaction of 32with hydrazine. The amino group was then modified to the corresponding iodide 34by standard diazotization and subsequent treatment with sodium iodide. Alkylation with 2‐fluorobenzylbromide mediated by cesium carbonate in DMF finally yielded iodide intermediate 21.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Contemporary Accounts in Drug Discovery and Development»
Представляем Вашему вниманию похожие книги на «Contemporary Accounts in Drug Discovery and Development» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Contemporary Accounts in Drug Discovery and Development» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.