Biosurfactants for a Sustainable Future
Здесь есть возможность читать онлайн «Biosurfactants for a Sustainable Future» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Biosurfactants for a Sustainable Future
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Biosurfactants for a Sustainable Future: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Biosurfactants for a Sustainable Future»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
A thorough introduction to the state-of-the-art in biosurfactant technology, techniques, and applications An exploration of biosurfactant enhanced remediation of sediments contaminated with organics and inorganics A discussion of perspectives for biomedical and biotechnological applications of biosurfactants A review of the antiviral, antimicrobial, and antibiofilm potential of biosurfactants against multi-drug-resistant pathogens. An examination of biosurfactant-inspired control of methicillin-resistant staphylococcus aureus Perfect for academic researchers and scientists working in the petrochemical industry, pharmaceutical industry, and in the agroindustry,
will also earn a place in the libraries of scientists working in environmental biotechnology, environmental science, and biomedical engineering.
Biosurfactants for a Sustainable Future — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Biosurfactants for a Sustainable Future», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
The soaps of Scheme 1.1show the most important structural characteristic of surfactants: the coexistence of one lyophilic group (alkyl chain) and one lyophobic group (carboxylate ion). In aqueous solutions, it is more frequent to use the terms hydrophilic and hydrophobic. A graphical representation head–tail (hydrophobic group–hydrophilic group) is widely used, the alkyl chain being the tail and the carboxylate group the head ( Figure 1.1). This structure gives the amphiphile character to surfactant compounds.
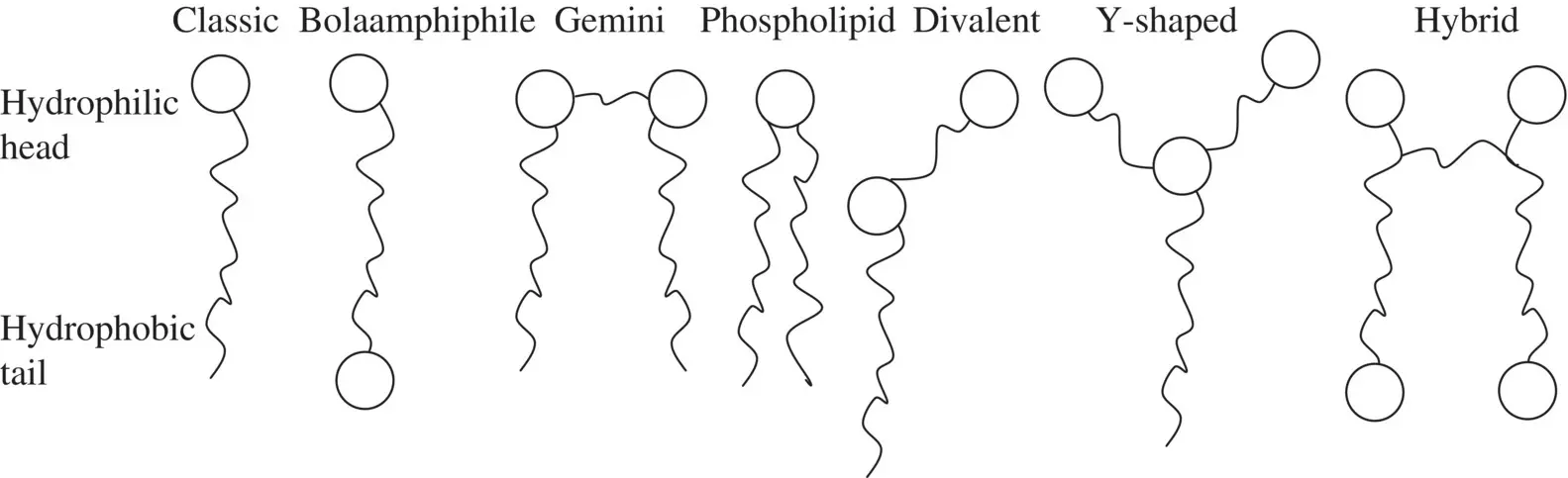
Figure 1.1 Schematic representation of the structure of some surfactants.
More generally, the head can be any polar group and the tail any apolar group, leading to a wide range of structures and types of surfactants. Among anionic heads, typical groups are carboxylate, sulfate, sulfonate, and phosphate, while the most frequent counterions are monovalent and divalent cations. Polycharged heads are also common, EDTA derivatives being well‐known examples [19]. Cyclopeptides constitute another important group [20]. Among cationic heads, typical groups are tetralkylammonium, N , N ‐dialkylimidazolinium and N ‐alkylpyridinium ions, while chloride and bromide are the most common counterions. Among neutral heads, polyethylene glycol ethers, polyglycol ethers, and carbohydrates can be mentioned. Zwitterionic heads are very important as phospholipids belong to this group, as well as sulfobetaines and trialkylamine oxides. Many examples can be found elsewhere [13].
However, the structures of surfactants may be more complex than the head–tail model suggests. For instance, the number of polar and non‐polar groups can be higher than one, the phospholipid phosphatidylcholine with two alkyl–allyl chains and a zwitterion as the head being an example. Gemini surfactants are dimeric surfactants [21] carrying two charged groups and two alkyl groups. The two amphiphilic moieties are connected at the level of the head groups, which are separated by a spacer group. They are characterized by critical micelle concentrations that are one to two orders of magnitude lower than those corresponding to conventional (monomeric) surfactants [22].
Bolaamphiphilic molecules contain a hydrophobic skeleton (e.g. one, two, or three alkyl chains, a steroid, or a porphyrin) and two water‐soluble groups on both ends [23]. They can be symmetric or asymmetric [24, 25]. Recent examples of bolaamphiphilic, Y‐shaped and divalent surfactants have been published by Baccile et al. [26] ( Figure 1.1).
Some surfactants, instead of the mentioned head–tail structure, present a bifacial polarity with the hydrophilic and hydrophobic characteristics at two opposite sides of the molecule. The best‐known examples are bile salts (see Figure 1.2) [27, 28]. Many membrane‐active compounds are facial amphiphiles including cationic peptide antibiotics [29]. The facial amphiphilic conformation adopted by these peptides is a consequence of their secondary and tertiary structures, allowing that one face of the molecule presents cationic groups (protonated amines or guanidines) and the other face contains hydrophobic groups. An example may be magainin I [30]. Among other surfactant structures, diblock copolymers and polymeric surfactants, fluorosurfactants and silicone‐based surfactants can be mentioned [13].
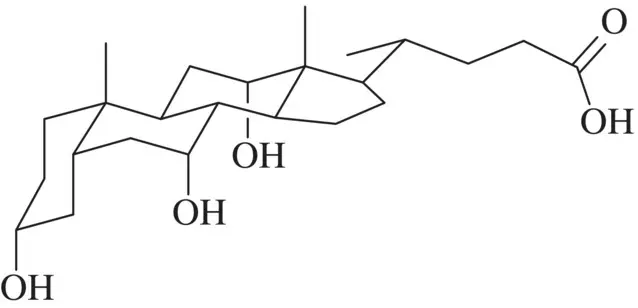
Figure 1.2 Bifacial structure of cholic acid.
1.2 Micelle Formation
The necessity of a quantitative measurement of the surface tension of soap solutions was soon evident. By the time that I. Traube published his earliest paper in 1884, significant theories of capillarity from La Place, Poisson, or Gauss were known [31]. Early measurements of the surface tension only imply inorganic salts, acids, and bases. In 1864 Guthrie [32, 33] measured some organic liquids. At the same time, Musculus [34] studied the capillarity of aqueous solution of alcohol observing that “the capillarity of the water decreases considerably with the addition of the least amount of alcohol, in the beginning, much faster than in the presence of more alcohol.” He also noticed that “all derivatives of ethyl alcohol which are soluble in water (as acetic acid) behave like this, and probably this is also the case with the other alcohols,” but substances such as “sugars, and salts if they are not present in a great amount, almost do not influence the capillarity of water.” He proposed the use of capillarity for measuring the concentration of alcohol and acetic acid in water, among other reasons, because “it offers the advantage that one needs only very little fluid for analysis, one drop being enough.” He continued that, as “the animal fluids, such as blood serum, urine, have a capillarity which is equal to that of water, it is possible to detect and quantify substances in the urine,” making reference, for instance, to bile.
Traube started the measurement of the influence of many organic substances on the surface tension of water in the period 1884–1885 [31] and observed that “the surface tension of capillary‐active compounds belonging to one homologous series decreased with each additional CH 2group in a constant ratio which is approximately 3:1,” leading him to propose Traube's Rule .
A nice historical paper was published by Traube [31] in 1940, in which he mentioned previous works related to the investigation of aqueous solutions of inorganic salts, acids, and bases, employing the method of capillary tubes, and, particularly, the dropping method applied by Quinke. Traube developed this method and designed a simple instrument, the stalagmometer – together with the stagonometer – which found general application in science and industry. In the mentioned paper, Traube refers mainly to his publications that appeared in the period 1886–1887. By 1906, the measurement of the surface tension by the capillary rise was so important that it was included in the book Practical Physical Chemistry by A. Findlay. The use of Traube's stalagmometer for such a purpose was proposed in the 3rd edition of the book, published in 1915. The experiment is still proposed in recent textbooks on practical Physical Chemistry [35].
Seventeen of the more important methods of measuring surface tension were described in 1926 by Dorsey [36]. According to his own words, “The list of references does not pretend to be complete but is intended merely to direct the reader to one or more of the sources from which the required information can be obtained most satisfactorily.” Even so, the number of cited papers was greater than 110, while the number of citations corresponding to the nineteenth century was 63 (56%). Eminent scientists such as Bohr, Rayleigh, Thomson, Kelvin, Maxwell, Laplace, and Poisson were among them. Tate [37] published his famous law in 1864 and Wilhelmy in 1863.
Even at low concentrations, surfactants reduce the surface tension of water due to its tendency to migrate toward the air–water interface, forming a monolayer. This was first suggested in 1907 by Milner [38] and, previously, Marangoni in 1871 “suggested that this capability [local variation in the tension of its surface] is due to the presence on the surface of the film of a pellicle, composed of matter having a smaller capillary tension than that of water.” Milner clearly established that “in several organic solutions the surface tension is less than that of water, and there is consequently an excess of solute in the surface.” Later, Langmuir [39] indicated that “the ‐COOH, ‐CO, and –OH groups have more affinity for water than for hydrocarbons… [and] when an oil is placed on water, the –COO– groups combine with the water, while the hydrocarbon chains remain combined with each other.” In other words, the tail of a surfactant (the hydrocarbon chain) must be located at the air interface, with the tail upwards oriented and the head (hydrophilic groups) at the water interface.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Biosurfactants for a Sustainable Future»
Представляем Вашему вниманию похожие книги на «Biosurfactants for a Sustainable Future» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Biosurfactants for a Sustainable Future» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.