Fundamentals of Pharmacology for Paramedics
Здесь есть возможность читать онлайн «Fundamentals of Pharmacology for Paramedics» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Fundamentals of Pharmacology for Paramedics
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Fundamentals of Pharmacology for Paramedics: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Fundamentals of Pharmacology for Paramedics»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
would also prove an indispensable resource for practicing paramedics seeking a practical, one-stop reference on a challenging subject.
Fundamentals of Pharmacology for Paramedics — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Fundamentals of Pharmacology for Paramedics», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Ion channels
Ion channels represent the only means for ions to cross cell membranes, and all cells contain multiple species of ion channel in their membranes. These channels can be gated in a number of ways, and drugs which can bind to specific channels can alter cellular activity profoundly by altering the passage of ions across the membrane, thereby altering the cell’s membrane potential. Most drugs that act in this way block ion channels rather than open them.
The local anaesthetic lidocaine, for example, acts by binding to and inhibiting voltage‐gated sodium channels in neuronal cell membranes, preventing the generation of action potentials by the affected neurons. Sensory neurons detecting touch, pressure and pain stimuli therefore become less responsive to those stimuli, resulting in anaesthesia.
The benzodiazepine class of drugs, including agents such as midazolam and diazepam, act by binding to a chloride ion channel in neuronal membranes.
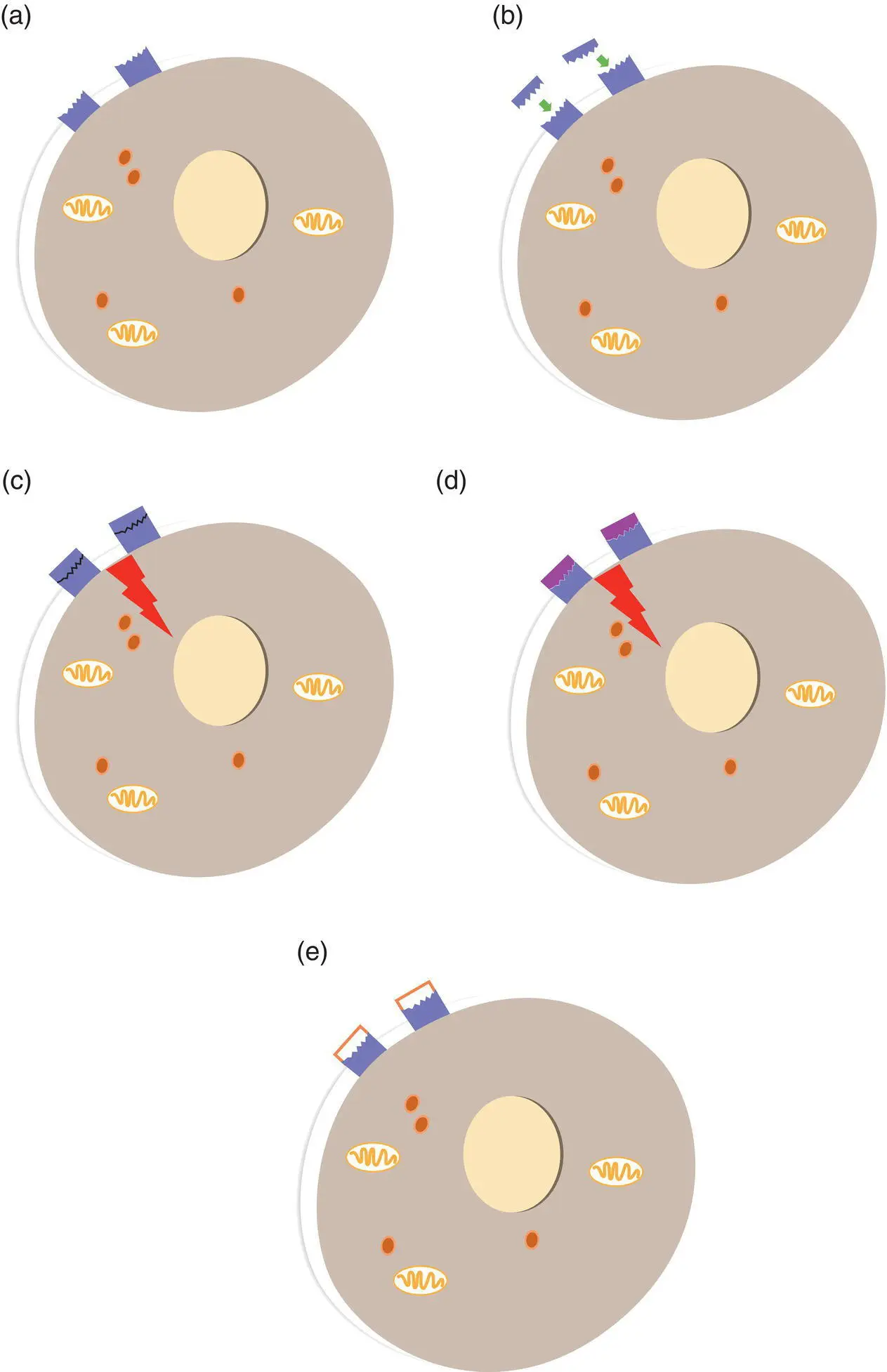
Figure 1.1 Drugs which act at receptors. (a) A cell has receptors for a specific signalling compound (e.g. a neurotransmitter or hormone) located on the cell membrane. (b) The endogenous signalling molecule binds to its receptors, fitting the receptor perfectly. (c) The binding triggers a series of actions inside the cell. These actions would be the normal response to that signalling compound. (d) If the molecular structure of a drug is sufficiently similar to that of the endogenous signalling compound, the drug will also be able to bind to the receptor and produce the same actions in the cell. This drug would be known as an agonist at this receptor. (e) If a drug has a molecular structure vaguely similar to that of the endogenous signalling compound, it may still be able to bind to the receptor, but not fit it perfectly enough to produce the same actions in the cell. This drug could prevent the endogenous signalling compound (and the agonist) getting to the receptor, thereby blocking their actions. This drug would be known as an antagonist at the receptor.
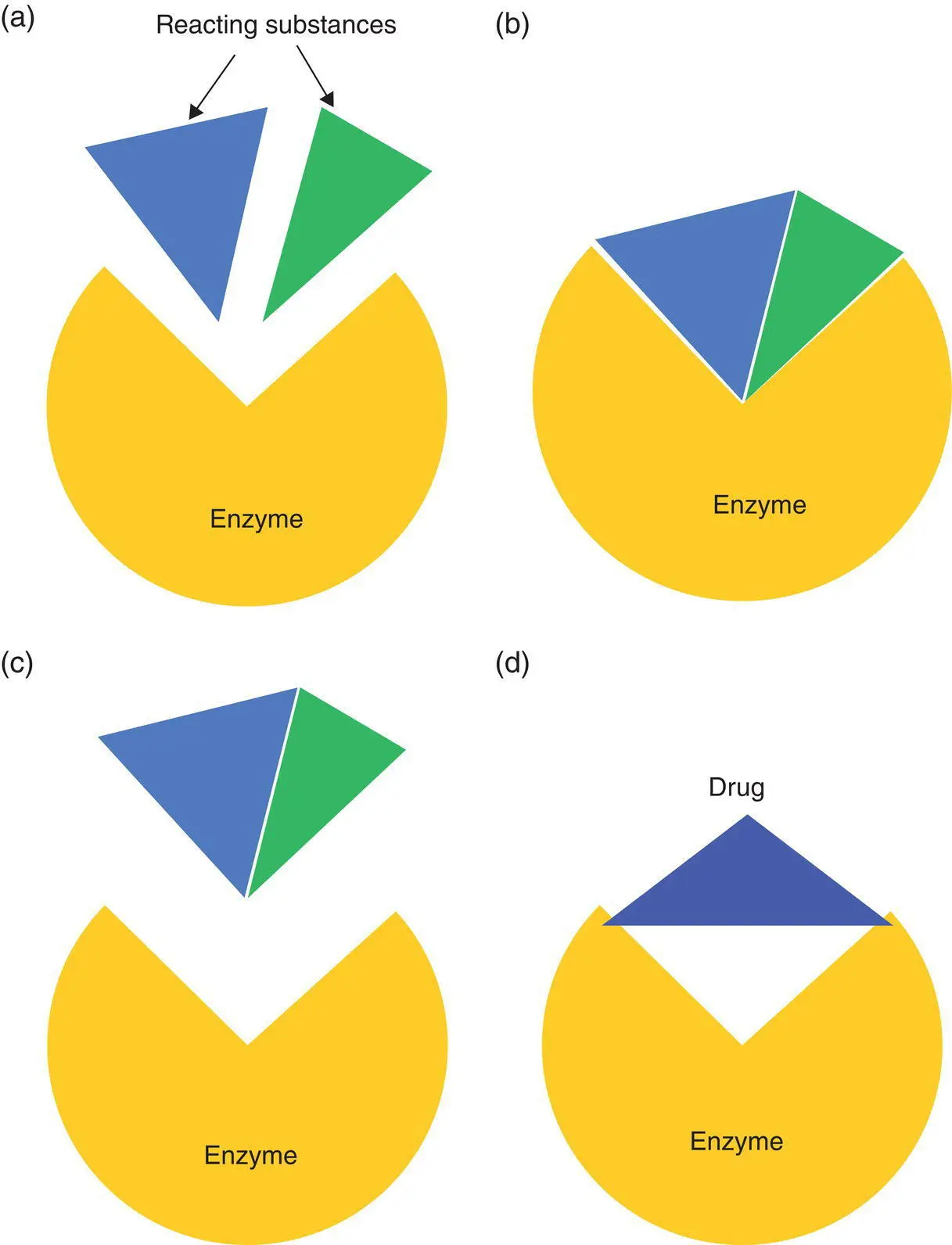
Figure 1.2 Enzymes operate by binding reacting substances (a) and accelerating their reaction – in this case the reaction is a combination of two molecules (b), then releasing the product from the enzyme’s binding site (c). A drug which can also bind to this site can prevent the catalytic function of the enzyme, thereby reducing the level of product (d).
This channel is opened normally by the inhibitory neurotransmitter GABA (gamma‐aminobutyric acid). Opening a chloride channel in the membrane allows the influx of negatively charged chloride ions to the cell, which hyperpolarises the cell, making it less likely to produce action potentials. The benzodiazepine class of drugs also act at this ion channel, albeit at a different site to GABA, but when they bind, they enhance the inhibitory actions of GABA, and add to the hyperpolarisation of neurons and the resulting nervous system depressant effect ( Figure 1.3).
Transport molecules
The large, complex proteins responsible for active transport of substances across cell membranes represent another valuable drug target for manipulation of physiological function.
There are active transporters or pumps in all cell membranes for sodium, potassium and calcium ions, and these are activated when those ions have to be transported across the cell membrane against their concentration gradient, i.e. from a lower to a higher concentration of ions. Ions can move through open ion channels if they are travelling down their concentration gradient, but will need active ‘pumping’ if they are to move the other way.
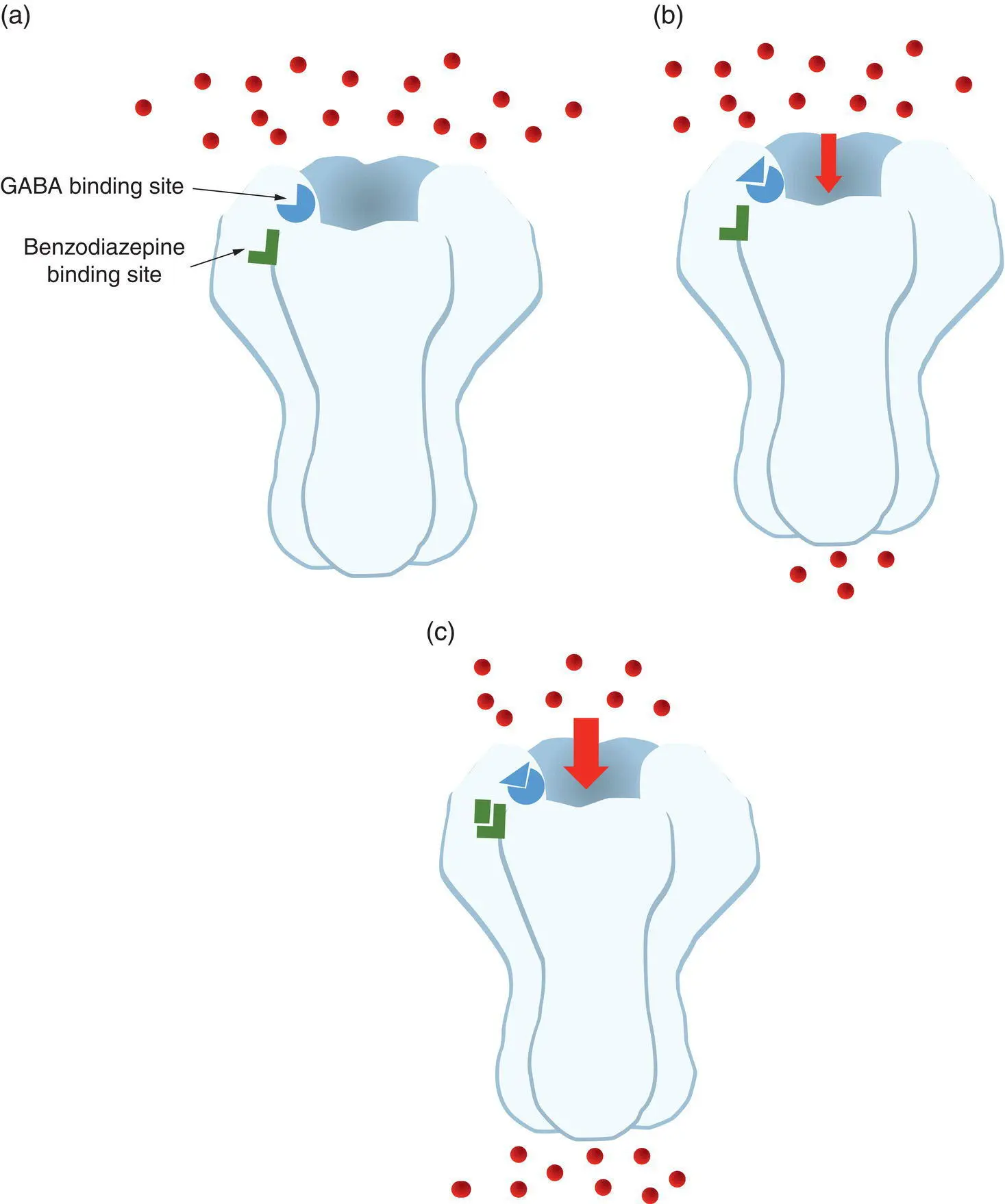
Figure 1.3 Benzodiazepines act by binding to a chloride channel. (a) The inhibitory neurotransmitter GABA has its receptor on the ligand‐gated chloride ion channel in neurons. The channel also has a binding site for drugs of the benzodiazepine class. (b) Binding of GABA to its receptor opens the channel, allowing chloride ions to flow into the neuron and hyperpolarise the membrane, inhibiting further neuronal activity. (c) Binding of a benzodiazepine to its site will, in the presence of GABA, further increase the flow of chloride ions into the cell by keeping the channel open for longer, thereby further inhibiting neuronal activity.
As with enzymes and ion channels, the drugs that bind transport molecules tend to inhibit the transport when they bind. This alters cell function by interfering with the distribution of ions across the cell membrane, thereby altering membrane potential. The cardiac glycoside drug digoxin, used to treat cardiac failure, acts by binding to a transport molecule – the sodium/potassium ATP‐ase or sodium/potassium pumps on the cell membranes of cardiac muscle cells. Digoxin inhibits the function of the sodium/potassium pumps, leading to an accumulation of sodium ions inside cardiac muscle cells, which in turn results in an accumulation of calcium ions in the cells (the additional intracellular sodium ions stimulate another transporter, which pumps sodium ions out of the cell in exchange for calcium ions). The increased level of calcium ions inside the cardiac muscle cells results in stronger contractions, which translates to a stronger, more forceful heartbeat.
Selectivity of binding and its effect
Some drugs are very selective in their binding sites, and can bind to a very limited number of sites, or only one site, but most drugs will be able to bind to more than one site. For example, a bronchodilator medication that acts as an agonist at adrenergic beta receptors may bind at only beta‐2 receptor subtypes, in which case it would be a selective beta‐2 agonist, but is more likely to bind to both beta‐1 and beta‐2 receptors, because of the degree of chemical similarity between the two receptor subtypes. The more selective a drug is for a single receptor, the fewer effects it is likely to bring about, so a more selective drug is likely to be one with fewer side‐effects. On the other hand, a less selective drug which activates two or three related receptors may have more therapeutic uses, but it will also have more side‐effects. The selectivity of a drug will tend to decrease as the dose increases, because binding to other receptor types will become more likely as the concentration of the drug increases. This helps to explain the dose dependency of many side‐effects of medications.
Clinical considerations
Salbutamol, also known as albuterol, is a beta‐2 receptor agonist and is frequently administered in the out‐of‐hospital setting for management of bronchospasm. It can also be used in the management of hyperkalaemia because it stimulates the transport of potassium ions from the blood into skeletal muscle cells. This effect is also mediated by the action of salbutamol on beta‐2 receptors.
Many patients who have been prescribed salbutamol may have already self‐administered their own ‘puffer’ prior to your arrival and may be tachycardic as a result. This is due to binding to beta‐2 receptors in cardiac muscle after absorption of salbutamol into the bloodstream. Tachycardia may predispose the patient to arrhythmias, so regarding these patients as high risk for a cardiac event is warranted.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Fundamentals of Pharmacology for Paramedics»
Представляем Вашему вниманию похожие книги на «Fundamentals of Pharmacology for Paramedics» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Fundamentals of Pharmacology for Paramedics» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.