Дмитрий Иванов - Нарушения обмена глюкозы у новорожденных детей
Здесь есть возможность читать онлайн «Дмитрий Иванов - Нарушения обмена глюкозы у новорожденных детей» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: Медицина, на русском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Нарушения обмена глюкозы у новорожденных детей
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Нарушения обмена глюкозы у новорожденных детей: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Нарушения обмена глюкозы у новорожденных детей»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Нарушения обмена глюкозы у новорожденных детей — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Нарушения обмена глюкозы у новорожденных детей», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Иммунная днзрегуляцня, полиэндокринопатия, энтеропатия, Х-связанная — IPEX-синдром.В 1996 году Peake J. Е. et al. [171] сообщили о мутации X хромосомы, сопровождающейся эксфолиативным дерматитом, диареей, гемолитической анемией, аутоиммунным поражением щитовидной железы и неонатальным диабетом. Дети умирают на первом году жизни от сепсиса. У некоторых детей описана агенезия островков Лангерганса. Аутоиммунная природа этого синдрома подтверждалась успешной терапией цитостатиками (Satake N. et al., 1993 [182]; Baud О. et al., 2001 [35]). Позже у больных были идентифицированы антитела к глутаминовой декарбоксилазе. Применение иммуносупрессивной терапии как этапа подготовки к трансплантации костного мозга приводило к купированию сахарного диабета, а затем диареи и дерматита. Установлено, что данная мутация, по крайней мере, в эксперименте, приводит к усиленной пролиферации СБ4+/СБ8-Т-лимфоцитов с мультиорганной инфильтрацией. В результате этого развивается гемофагоцитический синдром, и мальчики без лечения погибают на 15-25-й день жизни после рождения (Brunkow М. Е. et al., 2001) [47]. Описано три случая выздоровления после трансплантации красного костного мозга при данной патологии [35].
Синдром Wolcott-Rallison.Синдром назван в честь врачей Wolcott С. D., Rallison М. V. впервые в 1972 году, описавших трех больных в одной семье, имевших сочетание неонатального диабета и спондилоэпифизарных нарушений скелета [207]. Это аутосомно-рецессивное заболевание, характеризующееся ранним началом сахарного диабета, обычно в неонатальный период, и спондилоэпифизарными нарушениями роста. У больных часто отмечается гепатомегалия, задержка умственного развития, почечная недостаточность, ранняя смерть. В 2000 году Delepine М. et al. [66] описали двух подобных детей в разных семьях, имеющих мутацию 2р12. В данном локусе находится ген, являющийся регулятором синтеза инсулина и белков в β-клетках поджелудочной железы. В мире к 2010 году были описаны около 60 пациентов с синдромом Wolcott-Rallison (Julier С., Nicolino М., 2010) [116]. Хотя, конечно, больных небольшое количество, но, тем не менее, некоторые закономерности течения данного синдрома понятны. Сахарный диабет развивается в первые шесть месяцев жизни, спондилоэпифизарные нарушения роста начинают проявляться в первые два года жизни. Они выражены как клинически, так и рентгенологически (рис. 15–16).
Другие проявления более вариабельны, но среди них можно выделить: острую печеночную недостаточность, почечные дисфункции, экзокринную недостаточность поджелудочной железы, задержку ПМР, гипотиреоз, нейтропению, рецидивирующие инфекции. Достаточно часто отмечаются переломы костей.
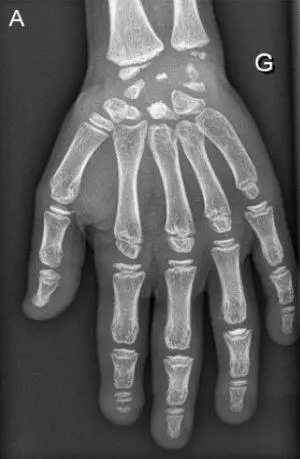
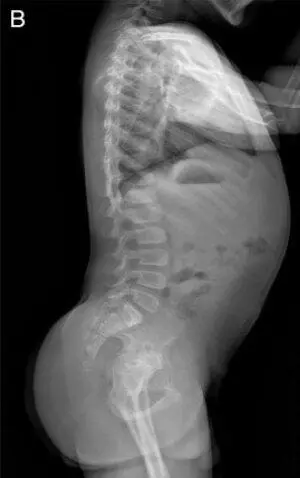
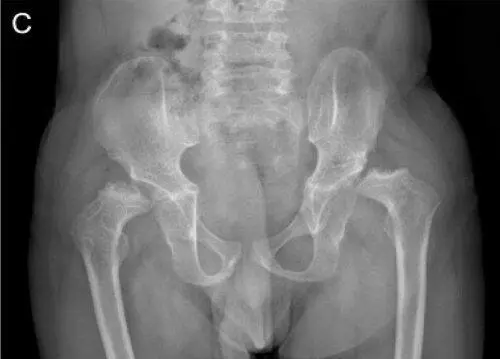
Рис. 15. Рентгенограммы пациента с синдромом Wolcott-Rallison (возраст 13 лет)
(Julier С., Nicolino М., 2010) [116].
А — руки: кости запястья небольшие с дисплазией дистального отдела лучевого и локтевого эпифизов; несколько фаланг являются диспластическими с аномальными укорочениями метафизов в проксимальном отделе. Б — позвоночник: в грудном отделе позвоночника имеются уплощения тел позвонков с дефектами переднего края. В — таз: гипоплазия вертлужной впадины с дисплазией эпифизов бедренных костей
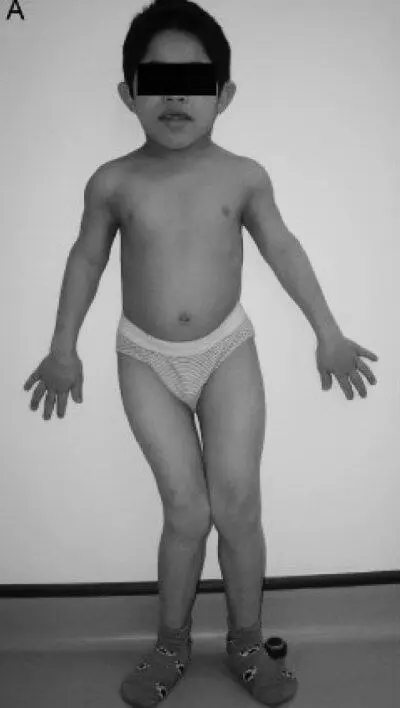
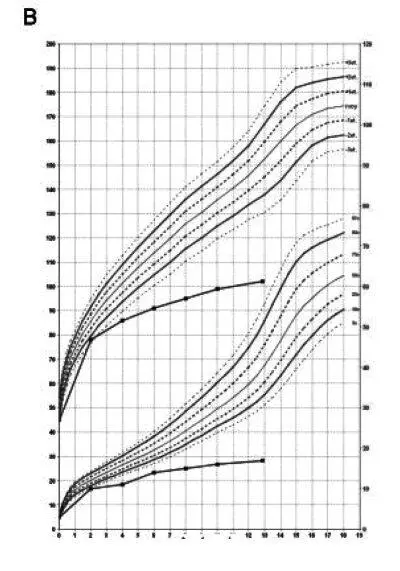
Рис. 16. Слева скелетные дисплазии и спондилоэпифизарные нарушения роста у пациента с синдромом Wolcott-Rallison (возраст 13 лет). Справа — диаграмма, демонстрирующая нарушения роста(Julier С., Nicolino М., 2010) [116]
Большая часть больных, рожденных от близкородственных браков, описана на Ближнем Востоке, Северной Африке, Пакистане и Турции. В настоящее время считают, что данный синдром должен быть исключен у любого ребенка, с диагностированным неонатальным сахарным диабетом (Rubio-Cabezas О. et al., 2009) [181]. Возможна генетическая и пренатальная диагностика данного синдрома (Stoy J. et al., 2007) [193]. Прогноз для жизни у больных неблагоприятный: описан только один больной, доживший до 35 лет (Senee V. et al, 2004 [185]; Ozbek М. N. et al, 2010 [168]). Christen H. J. et al. [52] в 1992 году описали двух мальчиков с Х-связанной высокой активностью фосфорибозил-АТФ-пирофосфатазой,развивших перманентный сахарный диабет в первый день жизни. Нарушенная толерантность к глюкозе сохранилась в течение жизни, хотя бывали периоды ремиссии, и больные не нуждались в инсулине. Оба ребенка кроме сахарного диабета имели задержку психического развития, атаксию и прогрессивную аксональную невропатию. Матери мальчиков страдали подагрой и гестационным диабетом.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Нарушения обмена глюкозы у новорожденных детей»
Представляем Вашему вниманию похожие книги на «Нарушения обмена глюкозы у новорожденных детей» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Нарушения обмена глюкозы у новорожденных детей» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.