Successful Drug Discovery, Volume 5
Здесь есть возможность читать онлайн «Successful Drug Discovery, Volume 5» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Successful Drug Discovery, Volume 5
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Successful Drug Discovery, Volume 5: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Successful Drug Discovery, Volume 5»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Successful Drug Discovery, Volume 5 — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Successful Drug Discovery, Volume 5», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
One sometimes finds what one is not looking for. When I woke up just after dawn on September 28, 1928, I certainly didn't plan to revolutionize all medicine by discovering the world's first antibiotic, or bacteria killer. But I suppose that was exactly what I did [29].
Streptomycin is another compound listed on the WHO list of essential medicines. It was isolated for the first time by Albert Schatz, a PhD student in the laboratory of Selman A. Waksman at Rutgers University in 1943. The results were published on 1 January 1944 [30], and the compound was quickly progressed to the clinics. Waksman, who also discovered several other important antibiotic natural products, among them actinomycin and neomycin, received the unshared Nobel Prize for medicine in 1952 “for his discovery of streptomycin, the first antibiotic effective against tuberculosis.” However, it is highly debated, if the role of other contributors, in particular of Schatz, was downplayed [31].
Gramicidin S was discovered by Georgyi Frantsevitch Gause, a Russian microbiologist and his wife in 1942 [32]. By 1943 it was being used to treat wounded Soviet soldiers in World War II. Gramicidin S is produced by Brevibacillus brevis and consists of two identical fivemers, which are coupled to give a cyclic decapeptide.

Figure 1.5 Lead structures isolated from natural sources.
1.4.2 Anticancer Drugs
1.4.2.1 Camptothecin
In 1952, the National Advisory Cancer Council discussed the promise of chemotherapy for curing cancer and came to the conclusion that the available knowledge was not sufficient to support establishment of a specific funding program for drug discovery for cancer chemotherapy. However, in 1955, the Congress of the United States approved foundation of the Cancer Chemotherapy National Service Center (CCNSC) [33] and an associated budget of US$ 5 million for research on cancer. US$ 4.2 million were dedicated to grants supporting specific research proposals, while US$ 800 000 were reserved for acquisition and testing of new compounds. As a consequence, dedicated profiling laboratories were set up and a large compound collection was compiled. This effort was even strengthened in 1960, when the National Cancer Institute (NCI) partnered with the US Department of Agriculture (USDA) to collect plant and animal samples in search for natural products with potential anticancer activities. This alliance turned out to be very productive. Between 1960 and 1981, a total of 30 000 compounds was screened, and many pharmaceutically interesting structures were identified. At one of the involved profiling laboratories, the newly founded Research Triangle Park in North Carolina, chemists Monroe Elliot Wall and Mansukh C. Wani reported, among many others, the structure and activity of the natural product called camptothecin ( Figure 1.6) [34].
Camptothecin, isolated from bark and stem of the Chinese Happy Tree ( Camptotheca ), was first chemically derived through total synthesis by Stork and Schultz [35] (Cornell University) in 1971, quickly followed by syntheses by the Danishefsky [36] (University of Pittsburgh) and Winterfeldt (University of Hanover) laboratories [37]. Camptothecin was identified as an inhibitor of topoisomerase I, acting through binding to the covalent topoisomerase‐DNA complex [38]. It is particularly toxic for cells in the S‐phase of mitosis. Albeit camptothecin itself proved too toxic to be used as a chemotherapeutic agent in patients, it served as a valuable lead structure for the approved drugs topotecan (Hycamtin™, approved in 1996 for treatment of ovarian cancer, in 2006 for cervical cancer, and 2007 for treatment of small‐cell lung carcinoma) and irinotecan (Camptosar™, a prodrug of topotecan approved in 1996 and used for treatment of colon cancer and small‐cell lung cancer) ( Figure 1.6). Both derivatives are derived through semisynthesis.
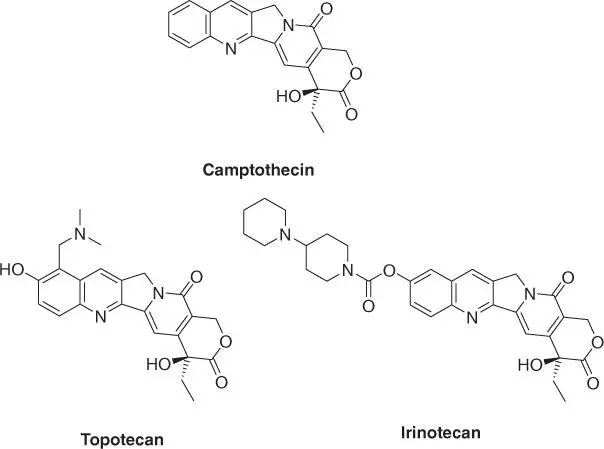
Figure 1.6 Camptothecin and approved derivatives.
1.4.2.2 Taxol
The discovery of Taxol™ (paclitaxel, Figure 1.7) is another success story resulting from this campaign. In 1962, USDA botanist Arthur Barclay was on an excursion in Gifford Pinchot National Forest in Washington State to collect samples for the screening campaign. Among another 200 samples collected over the course of several months, he chose to take needles, twigs, and bark of the pacific yew. This turned out to be an important moment in cancer drug discovery.
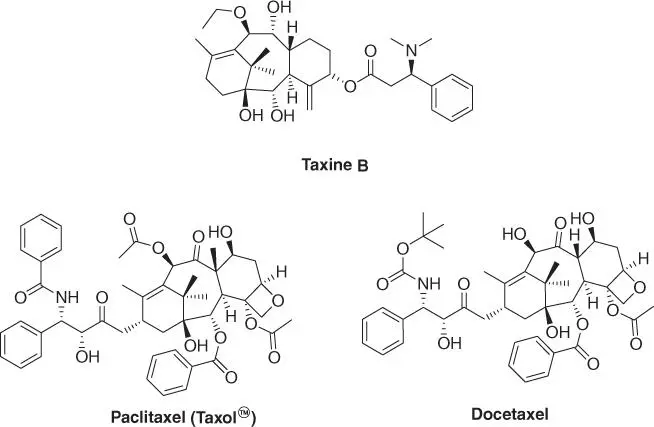
Figure 1.7 Taxol derivatives.
Two years later, Wall and Wani at Research Triangle Park, North Carolina discovered a promising anti‐leukemic and tumor inhibitory activity of an extract made from the collected stem bark [39]. However, the isolated yield from the dried bark was only 0.02 %. They contacted USDA and requested more material to supply further studies. In September 1964, Barclay went back to Gifford Pinchot National Park and collected another 30 lb of bark.
The yew tree itself has long been known to possess toxic properties. Almost any part of the tree is toxic but the red cup around the seeds is particularly hazardous. The lethal dose of needles of the common yew is estimated to be about 50 g for an adult. The toxic effects are caused by the contained taxine alkaloids (mainly taxine B), leading to cardiogenic shock [40]. These cardiac effects are distinct from the primary mechanism of action of Taxol and can be attributed to binding to ion channels. The main component of this activity seems to be taxine B ( Figure 1.7). Its structure is related to that of Taxol, but besides other differences, it lacks the oxetane ring and the benzoic amide and bears an exo ‐methylene group and a dimethyl amino residue. However, cardiotoxic side effects are also reported for paclitaxel.
Taxol did display interesting activities against various cell models of cancer and was moderately active in different models of leukemia. However, its solubility in aqueous media is very low. The initial overall interest in the compound was low, also as its availability was very limited. This changed quickly after new in vivo models were introduced at NCI in the early 1970s, and Taxol was found to be strongly active in a mouse model of melanoma. The pharmacological activity finally led to its nomination as a development candidate in 1977, triggering further examination.
In the same year, Susan Band Horwitz (Albert Einstein College of Medicine, Yeshiva University) was contacted by the NCI and was asked to explore the effects of Taxol [41]. She performed some initial experiments and observed that Taxol was capable of stopping replication of HeLa cells even at nanomolar concentrations due to its ability to induce mitotic arrest. Furthermore, she discovered a completely new phenotype. Cells treated with Taxol would be filled with stable microtubule bundles. In later research, it was determined that Taxol efficiently stabilizes microtubules, thus arresting cell cycle [42]. This new mechanism created a tremendous interest in Taxol. However, access to the compound was very limited. In fact, the bark of an estimated 3000 trees is needed to allow isolation of 1 kg of Taxol. Given that the tree will inevitably die after its bark is harvested and the pacific yew is a slow‐growing species, the development process was slowed down significantly.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Successful Drug Discovery, Volume 5»
Представляем Вашему вниманию похожие книги на «Successful Drug Discovery, Volume 5» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Successful Drug Discovery, Volume 5» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.