Genotyping by Sequencing for Crop Improvement
Здесь есть возможность читать онлайн «Genotyping by Sequencing for Crop Improvement» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Genotyping by Sequencing for Crop Improvement
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Genotyping by Sequencing for Crop Improvement: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Genotyping by Sequencing for Crop Improvement»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
A thoroughly up-to-date exploration of genotyping-by-sequencing technologies and related methods in plant science Genotyping by Sequencing for Crop Improvement,
Genotyping by Sequencing for Crop Improvement
Genotyping by Sequencing for Crop Improvement
Genotyping by Sequencing for Crop Improvement — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Genotyping by Sequencing for Crop Improvement», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
41 Vos, P.G., Uitdewilligen, J.G., Voorrips, R.E. et al. (2015). Development and analysis of a 20K SNP array for potato (Solanum tuberosum): an insight into the breeding history. Theoretical and Applied Genetics 128: 2387–2401. https://doi.org/10.1007/s00122‐015‐2593‐y.
42 Wang, S., Wong, D., Forrest, K. et al. (2014). International Wheat Genome Sequencing Consortium. Characterization of polyploid wheat genomic diversity using a high‐density 90 000 single nucleotide polymorphism array. Plant Biotechnology Journal 12: 787–796.
43 Whitcombe, D., Theaker, J., Guy, S.P. et al. (1999). Detection of PCR products using self‐probing amplicons and fluorescence. Nature Biotechnology 17 (8): 804–807.
44 Wickland, D.P., Battu, G., Hudson, K.A. et al. (2017). A comparison of genotyping‐by‐sequencing analysis methods on low‐coverage crop datasets shows advantages of a new workflow, GB‐eaSy. BMC Bioinformatics 18 (1): 586. https://doi.org/10.1186/s12859‐017‐2000‐6.
45 Winfield, M.O., Allen, A.M., Burridge, A.J. et al. (2016). High‐density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant Biotechnology Journal 14 (5): 1195–1206.
46 Zhang, P., Li, X., Gebrewahid, T.W. et al. (2019). QTL mapping of adult‐plant resistance to leaf and stripe rust in wheat cross SW 8588/Thatcher using the wheat 55K SNP array. Plant Disease 103 (12): 3041–3049.
47 Zhao, K., Tung, C.W., Eizenga, G.C. et al. (2011). Genome‐wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nature Communications 2 (1): 1–10.
3 Opportunity and Challenges for Whole‐Genome Resequencing‐based Genotyping in Plants
Surbhi Kumawat1, Gaurav Raturi1, Pallavi Dhiman1, Sreeja Sudhakarn1, Nitika Rajora1, Vandana Thakral1, Himanshu Yadav1, Gunashri Padalkar1, Yogesh Sharma1, Vinaykumar Rachappanavar2, and Manish Kumar2
1 Department of Agriculture Biotechnology, National Agri-Food Biotechnology Institute (NABI), Mohali, Punjab, India
2 Department of Seed Science and Technology, Dr. Yashwant Singh Parmar University of Horticulture and Forestry, Solan, Himachal Pradesh, India
3.1 Introduction
Recent advancements in next‐generation sequencing (NGS) have speed up genomics and transcriptome research across the board, especially in plant science (Rana et al. 2019; Ratnaparkhe et al. 2020). Genomics is a key for the better understanding of whole‐genome involving the DNA sequences determined in the order in which they are present in a genome which indeed in the sequential exploration of the evolution of genome structure and interpretation of the molecular phylogeny. It involves the combination of various technologies including recombinant DNA, sequencing methods of DNA, and bioinformatics to assemble, analyze and assign the structure and function of the plant genome. Earlier, the Sanger method was commonly used for nucleic acid sequencing in genomics but in 2005 the development of NGS brought sequencing platforms that are fast and cost‐effective enabling the excavating of characteristics unique to particular species in terms of gene structure as shown in Figure 3.1(Unamba et al. 2015; Loera‐Sánchez et al. 2019). It also enables us to understand the mechanism lying behind the process of gene expression as well as secondary metabolism. Further recent advances in the NGS platforms bring the gratifying efforts in de‐novo whole‐genome/transcriptomic sequencing and also for the developments of markers based on genome‐wide sequences (Vasupalli et al. 2020). The most common platforms of NGS include Illumina/Solexa, ABI/SOLiD, Helicos, and 454/Roche (Unamba et al. 2015).
Earlier techniques for gene expression analysis include northern blotting in which specific RNA are detected and quantified (Goda and Minton 1995). Another technique involves the hybridization of antisense RNA with the complementary known target sequence thereby preventing the digestion of target sequence by single‐strand RNAase resulting in the specific and accurate quantification of the target sequence. But the major drawbacks associated with this technique include it requires prior knowledge about the target sequences. With the advancements in technologies, transcriptomics came into existence which analyses all RNA molecules (tRNA, rRNA, mRNA, and other noncoding RNA) transcribed in an organism. Microarray is a widely employed technique to analyze the transcriptome. In a single experiment, this technique can measure the expression level of a thousand genes but there are several issues associated with its sensitivity and reproducibility.
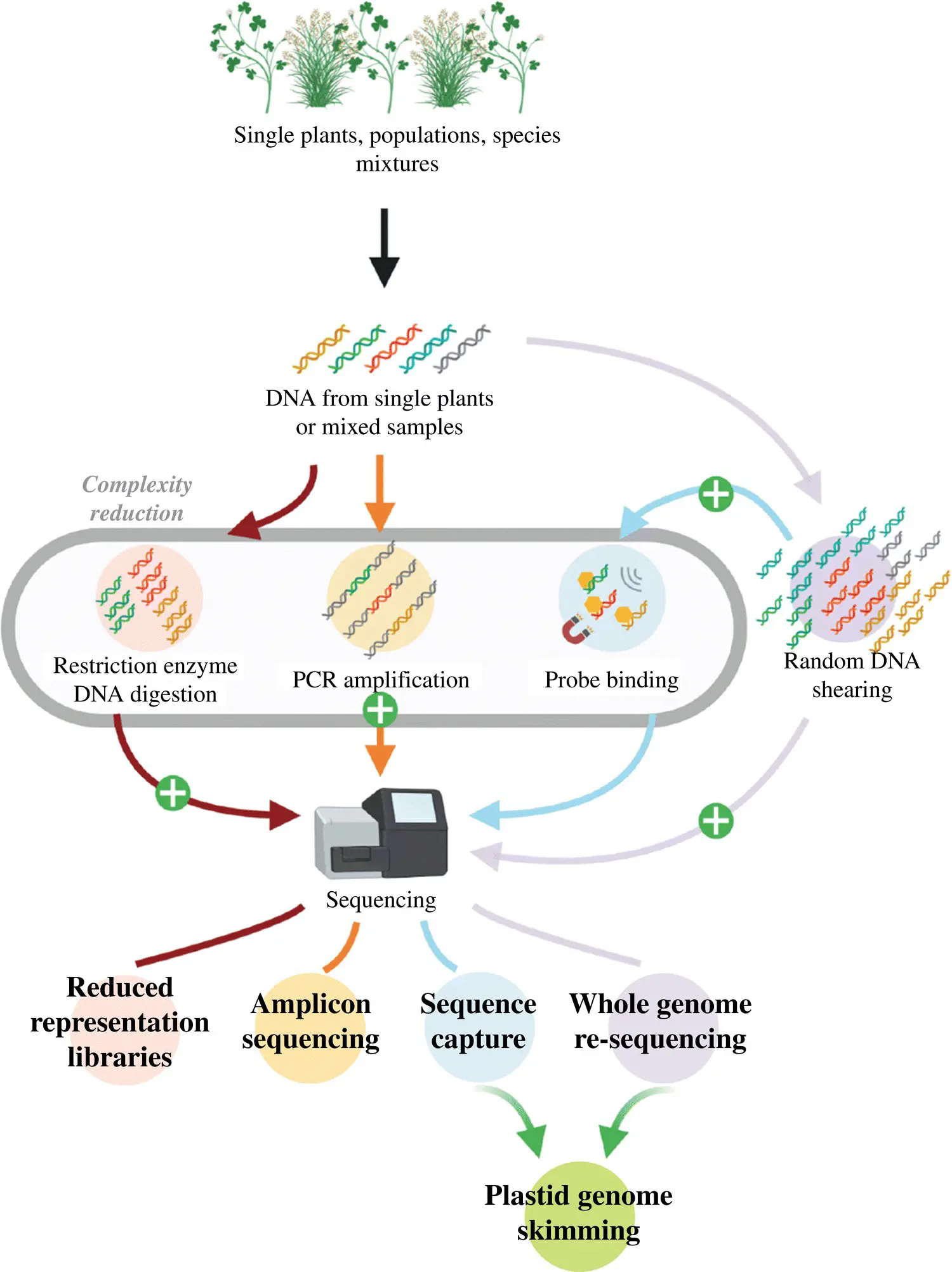
Figure 3.1 Diagrammatic representation of various high‐throughput‐sequencing methods used in assessments of genetic diversity. Methods mentioned in grey oval represent the complexity reduction. Library preparation steps are indicated by plus sign. Red arrow indicates reduced representation libraries produces by starting DNA sample digestion with restriction enzyme. Orange arrow indicates amplicon sequencing while blue arrow represents the sequence capture. Whole‐genome resequencing is indicated in purple arrow in which random fragments of digested DNA are sequenced. Green arrow represents plastid genome skimming responsible for plastid genome sequences recovery.
The figure is reproduced from Loera‐Sánchez et al. (2019) which is available under a Creative Commons Attribution 4.0 (CC‐By 4.0) International License, which permits reproduction.
The recent advancement in NGS includes next‐generation RNA sequencing (RNA‐seq) (Sharma et al. 2011; Sonah et al. 2016). This technique mainly focuses on the mRNA sequencing of only those genes that are expressed in the transcriptome (Chaudhary et al. 2019b). This technique helps in the identification of novel genes by de novo assembly without reference genome mapping. NGS also aids in the development of molecular markers. NGS has led to the exploration of thousands of markers on the entire genome resulting in an ease in the genome‐wide association studies. These markers also aid in association mapping (Sonah et al. 2015; Zargar et al. 2015). These technologies have enabled us to understand the underlying process of gene expression and the development of resources for the analysis of marker‐assisted breeding (MAS) and diversity analysis (Unamba et al. 2015).
3.2 Basic Steps Involved in Whole‐Genome Sequencing and Resequencing
Whole‐genome sequencing (WGS) can be divided into two groups, which include de novo WGS and whole‐genome resequencing (WGR) (Bhat et al. 2020). WGS involves the genome sequence assembly for the first time while WGR compares genomic variability within individuals or populations (Patil et al. 2019). WGR requires the prior availability of reference genome for mapping and variant detection. Among WGS, de novo WGS involves the complete assembly of a species genome for the first time (Sevanthi et al. 2018). First, for the library preparation, high quality of genomic DNA is subjected to fragmentation followed by the addition of adaptors to the DNA fragments. For the detection of small structural variants such as INDELs or CNVs (copy number variations), short reads (350–550 bp insert size) from standard libraries are utilized while long‐read data or mate‐pair libraries with an insert size of around 2 to 20 kb will be required for the detection of large structural variants. For high‐throughput sequencing, Illumina is often used. The sequences are mapped on the genome sequence based on similarity and local contigs are developed. While assembling the sequence, repetitive regions show difficulty in alignment with the short reads. In that case, mate pair‐end sequencing reads aids in aligning large sequences which are also referred as scaffolds or supercontigs by linking and orienting contig. Unknown sequences gaps are denoted as Ns. The final result of a genome assembly corresponds to the contiguous scaffold sequences in a series separated by gaps.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Genotyping by Sequencing for Crop Improvement»
Представляем Вашему вниманию похожие книги на «Genotyping by Sequencing for Crop Improvement» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Genotyping by Sequencing for Crop Improvement» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.