Дж. Йео - Прожорливый ген. Диеты и лишний вес с точки зрения генетики
Здесь есть возможность читать онлайн «Дж. Йео - Прожорливый ген. Диеты и лишний вес с точки зрения генетики» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. ISBN: , Жанр: Прочая научная литература, Биология, на русском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Прожорливый ген. Диеты и лишний вес с точки зрения генетики
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:978-5-17-114228-5
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Прожорливый ген. Диеты и лишний вес с точки зрения генетики: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Прожорливый ген. Диеты и лишний вес с точки зрения генетики»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
В эпоху постправды, когда сориентироваться в информационном потоке необычайно сложно, Йео помогает разобраться в человеческой физиологии и гормональных функциях и указывает, в какой мере энергоемки различные физиологические процессы. Уложенное в логичную структуру знание поможет достигнуть гармонии в отношениях с едой, телом и весом.
В формате PDF A4 сохранен издательский макет книги.
Прожорливый ген. Диеты и лишний вес с точки зрения генетики — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Прожорливый ген. Диеты и лишний вес с точки зрения генетики», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
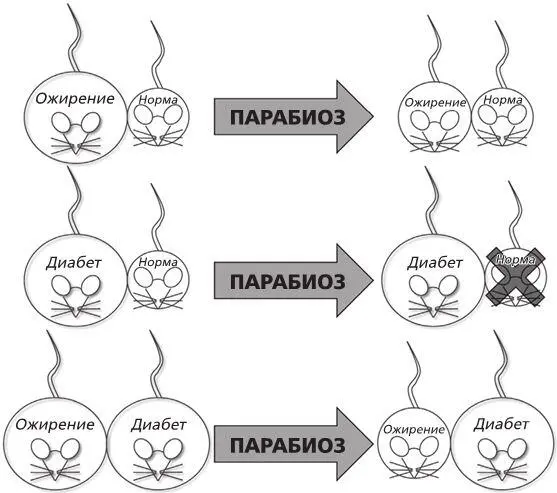
Рисунок 4. Парабиотические эксперименты Дага Коулмена, которые привели к появлению гипотезы о гормоне сытости.
Провели три эксперимента. В первом ученый соединил мышь с ожирением и нормальную мышь. Он увидел, что с нормальной мышью ничего не случилось, а толстая сильно похудела. Во втором он соединил мышь, больную диабетом, и нормальную. На этот раз нормальная мышь перестала есть, потеряла вес и в конце концов умерла, а с больной ничего не случилось: она продолжала есть и не теряла веса. В третьем эксперименте «сиамскими близнецами» оказались мышь с ожирением и мышь с диабетом. Как и в первом случае, мышь с ожирением перестала есть и стала терять вес, а мышь с диабетом вела себя так же, как и до эксперимента [12] Coleman, D. L., ‘Effects of parabiosis of obese with diabetes and normal mice.’ Diabetologia 9 (1973), 294–8.
. Все это было очень странно.
Тем не менее самой очевидной частью эксперимента стало то, что мышь с ожирением и мышь с диабетом страдали от мутаций в разных генах, потому что они вели себя по-разному, когда их соединяли либо с мышью нормального размера, либо друг с другом. Не такой очевидной (по крайней мере, для меня) оказалась идея, которую высказал Даг. Он предположил, что у мыши с ожирением не хватало «маркера сытости» – назовем его так, – это вещество находится в кровеносной системе, и когда его количество растет, мышь прекращает есть. У мыши с диабетом проблем с «маркером сытости» не было, только она не могла на него реагировать. Удивительное заключение, конечно. Но вот какая тут была логика: когда мышь с ожирением была присоединена к нормальной мыши, она неожиданно стала получать сигнал о том, что наелась, потому что этот сигнал присутствовал в кровообращении нормальной мыши. В результате она прекратила есть слишком много и стала терять вес. Во втором эксперименте мышь с диабетом была к этому фактору нечувствительна: ее тело производило огромные количества этого вещества, так что когда нормальная мышь получала его вместе с кровью, она немедленно прекращала есть. Третий эксперимент, когда были соединены мыши с диабетом и ожирением, доказал, что мутировавшие гены двух мышей взаимодействовали друг с другом, потому что высокий показатель насыщения у мыши с диабетом передался мыши с ожирением, и тогда она прекратила есть.
Такова, по крайней мере, была гипотеза. И теперь надо было проверить еще одну маленькую деталь.
Только в 1980-х годах бурное техническое развитие в области генетики позволило команде из Рокфеллеровского университета в Нью-Йорке начать исследование, которое действительно позволило продвинуться вперед в проверке этой гипотезы. В 1994 году, через 45 лет после того, как в одной из лабораторных клеток была замечена мышь с ожирением, лаборатория Джеффри Фридмана сообщила, что у таких мышей происходит мутация в гене, который Джефф решил назвать лептином [13] Zhang, Y. et al., ‘Positional cloning of the mouse obese gene and its human homologue.’ Nature 372 (1994), 425–32, doi:10.1038/372425a0.
.
Лептин, как выяснилось, производится в жире и затем поступает в кровь, где циркулирует как гормон. Когда Джефф получил лептин и впрыснул его мышке с ожирением, она перестала есть и похудела, подтвердив таким образом, что раньше у нее был недостаток нормального лептина. Это и был таинственный «гормон сытости», о котором говорил Даг. Еще через пару лет у мыши с диабетом была найдена мутация в рецепторе лептина, потому-то она и не худела, когда ей давали лептин [14] Chen, H. et al., ‘Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice.’ Cell 84 (1996), 491–5.
. И опять-таки предположение Дага – о том, что в крови у этой мыши был лептин, но она не могла его правильно воспринимать, – оказалось поразительно точным.
То, что ученые обнаружили лептин и его рецептор, впервые указало на присутствие в организме такой гормональной системы, которая может регулировать прием пищи и ничего не имеет общего с силой воли. Но затем появился еще один важный вопрос. Присутствует ли эта странная система только у мышей или же ею обладают и другие млекопитающие? Чтобы ответить на этот вопрос, в начале 1997 года к исследованиям подключились мой старый коллега Стивен О’Райли и его (в то время) студентка Садаф Фаруки. Как мы помним, приводившая к ожирению мутация в гене лептина не была создана искусственно, она получилась сама собой. Стивен – усатый ирландец, который любит поразмышлять. И в ходе своих размышлений он пришел к такому предположению: если мутация в гене могла привести к ожирению у млекопитающего, вероятно, подобные случаи ожирения есть и у людей. В одной из лабораторных морозильных камер у Стивена имелись пробирки с образцами крови двоих детей – они были двоюродными братом и сестрой, оба страдали ожирением. Садаф попыталась измерить лептин в этих образцах, но не смогла. Вернее сказать, анализ не показал присутствия лептина. Тогда Карл Монтагю, который проходил у Стивена постдокторантуру, определил строение гена лептина у этих детей и выявил мутацию – отсутствие одного-единственного гуанина (помните четыре элемента, из которых собрана ДНК: аденин (А) тимин (Т), гуанин (Г) и цитозин (Ц)). Нехватка гуанина была причиной того, что у детей в организме отсутствовал правильно функционирующий лептиновый белок. Стив в очередной раз оказался прав. Его команда в Кембриджском университете обнаружила – впервые в истории генетики, – что мутация в одном гене может привести к тяжелым формам ожирения у людей [15] Montague, C. T. et al., ‘Congenital leptin deficiency is associated with severe early- onset obesity in humans.’ Nature 387 (1997), 903–8, doi:10.1038/43185.
.
Интервал:
Закладка:
Похожие книги на «Прожорливый ген. Диеты и лишний вес с точки зрения генетики»
Представляем Вашему вниманию похожие книги на «Прожорливый ген. Диеты и лишний вес с точки зрения генетики» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.




![Олеся Галькевич - Тараканы в твоей голове и лишний вес [publisher - SelfPub]](/books/385885/olesya-galkevich-tarakany-v-tvoej-golove-i-lishnij-v-thumb.webp)







Обсуждение, отзывы о книге «Прожорливый ген. Диеты и лишний вес с точки зрения генетики» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.