Albert P. Li - Transporters and Drug-Metabolizing Enzymes in Drug Toxicity
Здесь есть возможность читать онлайн «Albert P. Li - Transporters and Drug-Metabolizing Enzymes in Drug Toxicity» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Transporters and Drug-Metabolizing Enzymes in Drug Toxicity
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Transporters and Drug-Metabolizing Enzymes in Drug Toxicity: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Transporters and Drug-Metabolizing Enzymes in Drug Toxicity»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Unites both the metabolism and transporter components of drug toxicity – two aspects not normally connected and the latter often neglected Familiarizes readers with the mechanism and species differences in drug metabolizing enzymes and transporters Discusses promising approaches to accurately predict human drug toxicity via the incorporation of human drug metabolism in toxicity evaluation
Transporters and Drug-Metabolizing Enzymes in Drug Toxicity — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Transporters and Drug-Metabolizing Enzymes in Drug Toxicity», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
2.3.2 Dose and Reactive Mtabolites
The idiosyncratic nature of DILI suggests that it is independent of dose; however, many DILI cases have occurred with doses of drugs >100 mg per day, and cases for drugs given at doses of 10 mg per day or lower have been reported only rarely [13]. Furthermore, the average daily dose of drugs reported to cause hepatotoxicity was higher compared to drugs not associated with this [13]. Moreover, extensive hepatic metabolism has been associated with a higher risk of hepatic adverse events from oral medications [14]. High daily dose and extensive drug metabolism in the liver are associated with high risk of DILI, supporting the hypothesis that the formation and accumulation of RMs beyond a critical threshold is the precondition triggering the development of liver injury.
The quantitative relationship between daily dose, formation of RMs and risk of DILI in humans was reported by Chen et al. [15]. By considering N = 354 FDA‐approved oral medications, the authors defined an algorithm for assigning a DILI score by factoring the relative contribution of daily dose, logP and RM, which permitted a quantitative assessment of clinical DILI risk:

The three parameters in the DILIscore formula (i.e. daily dose/Cmax, logP, and RM formation) contributed significantly to DILI risk with the order of RM > daily dose > logP. The relationship between calculated DILI scores and DILI risk was validated by three independent datasets retrieved from the literature, and the score model demonstrates its correlation with the severity of clinical outcome by applying to N = 159 clinical cases collected from the NIH’s LiverTox database ( https://www.ncbi.nlm.nih.gov/books/NBK547852/).
2.3.3 Structural Alerts for Avoiding Reactive Metabolites
A compound’s potential to form electrophiles and chemically RMs is determined by its chemical structure. Certain functional groups, referred to as structural alerts, are molecular fragments with high chemical reactivity, or which can be transformed into fragments with high chemical reactivity through bioactivation [16]. These structural alerts can be present in the parent compound or in its metabolites.
Structural alerts regularly have been employed for screening potential RM formation from candidate compounds in drug discovery, in order to limit undesirable toxic effects. General avoidance of certain functional groups related to RM formation is considered a normal practice at the lead optimization/candidate selection stage. Sometimes, if the structural alert must be carried into lead optimization, mitigation strategies learned from industry experience may be suggested to attenuate or avoid toxicity, including: (i) use substitutes resistant to metabolism to replace potential structural alerts; (ii) decrease electronic density; and (iii) introduce a structural element to redirect metabolism potential [16].
An increasing concern, however, is that the value of incorporating structural alerts for screening drug molecule‐associated safety hazards may have been overstated. In a study examining the role of RMs, about 50% of the top 200 drugs marketed in the U.S. and ranked by prescription and sales were found to have one or more alerts in their chemical architecture [17]. Meanwhile, the absence of structural alerts cannot serve as a guarantee of drug safety. For example, ximelagatran did not present any alerts in its chemical structure but was recalled as a thrombin inhibitor with idiosyncratic hepatotoxicity [17].
Figure 2.3lists some typical structural alerts found in the literature. Since the relationships among these alerts in generating RMs and toxicity are largely unclear, most institutes and companies maintain their own libraries of structural alerts. Several review papers have summarized a comprehensive list of alerts. Enoch et al., for example, reported many structural alerts, each with an outline of their associated mechanistic chemistry of adduct formation. These alerts have been incorporated into the Organization for Economic Co‐operation and Development (OECD) (Q)SAR Toolbox, a freely available software tool for analyzing structural alerts in the public domain [18]. Claesson and Minidis summarized 63 alerts with detailed SMART names, SMART pictures and examples of drugs, which could be the basis for an in‐house structural alert application system. [19]
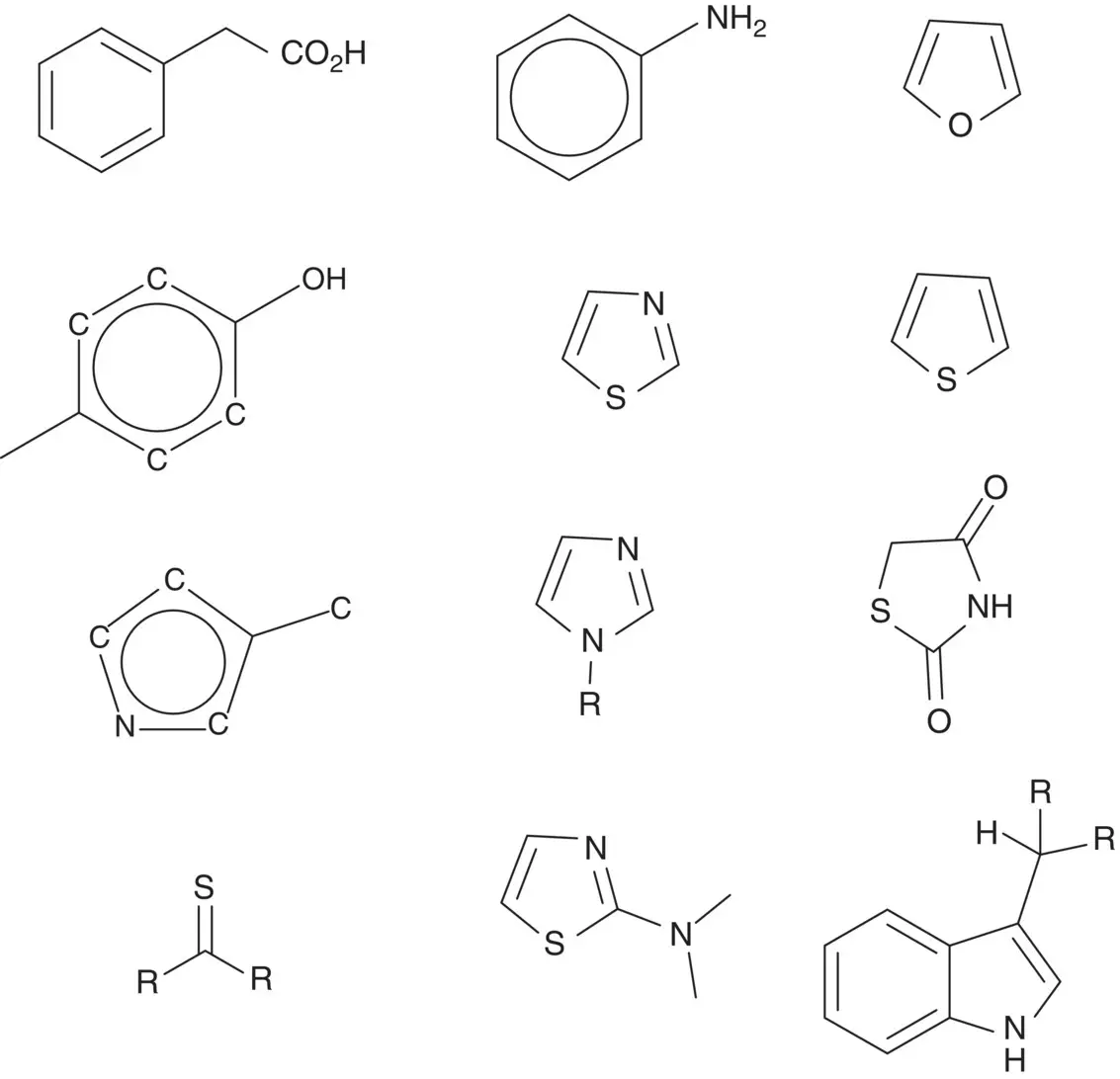
Figure 2.3 Some typical structure alerts for formation of reactive metabolites found in the literature.
2.3.4 Experimental Approaches for Assessing Reactive Metabolites
The structural alerts for formation of RMs sometimes do not imply actual formation of RMs. In this circumstance, experimental evidence is needed to assess whether a drug candidate is bioactivated to RMs. We discuss some selected experimental approaches here, including covalent binding assay, electrophile trapping experiment and time‐dependent inactivation of CYP450.
2.3.4.1 Covalent Binding Assay
The earliest approach for assessing protein covalent binding is using radiolabeled compounds with human liver microsomal or hepatocyte incubations. It is the gold‐standard approach for measuring RM formation and can quantify the extent of RMs covalently binding to proteins. The original experimental protocol for determining covalent binding properties of radioactive drugs was developed by Evans et al. [20]. In their protocol in vitro human liver microsomes are used as the metabolizing enzyme source and incubated with radioactive drug and cofactors for oxidative metabolism. Quantification of covalent binding is measured through counting the radioactivity of the protein pellet.
Although the covalent binding assay is reliable and widely used by pharmaceutical companies, the use of radiolabeled drugs makes running high throughput screening difficult, and it is an expensive option for assessing whether a compound is likely to undergo bioactivation. A more common and economical screening approach is using electrophile trapping experiments that do not require radiolabeled drugs.
2.3.4.2 Electrophile Trapping Experiments
Electrophile trapping is the primary approach for screening RMs in the early phase of drug discovery without involving a radioactive drug. Normally it requires the addition of trapping agents (i.e. GSH) to microsomal incubation followed by liquid chromatography mass spectrometry (LC‐MS) analysis. This approach also helps indicate which metabolite structure is responsible for this reactivity and characterizes the mechanism of covalent binding. It generates the stable trapping adducts and/or conjugates of electrophilic RMs using nucleophilic trapping reagents or in vitro incubation, along with hepatic microsome and cellular, animal, and human studies. The biological nucleophile GSH is the most frequently‐used nucleophile reagent, commonly used for trapping soft electrophiles (e.g. epoxides, quinones, and quinone methides). For certain hard electrophiles with high charge density and high polarization, hard nucleophiles such as cyanide anion (CN −) are preferred because the GSH adducts are unstable and GSHs have limited trapping efficiency.
Following in vitro incubations, various analytical approaches such as LC‐MS, fluorescence or radiochemical detection could be used to analyze the trapped adducts and/or conjugates of RMs. Mass spectrometric methods are the most common approach. Taking advantage of specific fragmentation behavior of the peptide moiety of nucleophilic trapping reagents, stable trapping adducts, and/or conjugates of electrophilic RMs can be detected and characterized. For example, the neutral loss of 129 Da is commonly used by positive ion electrospray‐tandem mass spectrometry to provide a generic endpoint for GSH trapping and measurement. Without using the radiolabeled drugs, this protocol can provide semi‐quantitative estimates of adduct formation for RMs.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Transporters and Drug-Metabolizing Enzymes in Drug Toxicity»
Представляем Вашему вниманию похожие книги на «Transporters and Drug-Metabolizing Enzymes in Drug Toxicity» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Transporters and Drug-Metabolizing Enzymes in Drug Toxicity» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.