Albert P. Li - Transporters and Drug-Metabolizing Enzymes in Drug Toxicity
Здесь есть возможность читать онлайн «Albert P. Li - Transporters and Drug-Metabolizing Enzymes in Drug Toxicity» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Transporters and Drug-Metabolizing Enzymes in Drug Toxicity
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Transporters and Drug-Metabolizing Enzymes in Drug Toxicity: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Transporters and Drug-Metabolizing Enzymes in Drug Toxicity»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Unites both the metabolism and transporter components of drug toxicity – two aspects not normally connected and the latter often neglected Familiarizes readers with the mechanism and species differences in drug metabolizing enzymes and transporters Discusses promising approaches to accurately predict human drug toxicity via the incorporation of human drug metabolism in toxicity evaluation
Transporters and Drug-Metabolizing Enzymes in Drug Toxicity — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Transporters and Drug-Metabolizing Enzymes in Drug Toxicity», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
3.8.1 Drug Metabolism and Toxicity
As for most marketed drugs that are found to be associated with idiosyncratic liver toxicity, Sitaxentan has been extensively evaluated in preclinical safety tests in multiple animal species, including single‐ and multiple‐dose studies in mice, rats, and dogs. Preclinical findings were non‐remarkable. While signs of liver toxicity including liver hypertrophy were observed, the preclinical safety study results show that Sitaxentan has acceptable safety margins [158].
As of this writing, there is only one study on the metabolic fate of Sitaxentan. Using in vitro hepatic experimental systems including human hepatocytes, human liver microsomes, and complementary DNA (cDNA) expressed microsomes, the 1,3‐benzodioxole ring of Sitaxentan is found to undergo enzymatic demethyleneation to an ortho‐catechol metabolite that can further oxidize to a reactive ortho‐quinone metabolite which undergoes GSH conjugation. CYP3A4 is found to be the major P450 isoform involved in the metabolic activation of Sitaxentan and is also inactivated during the metabolic process, further confirming the formation of highly reactive metabolites. Metabolic activation of Sitaxentan by CYP3A4 to reactive quinone metabolites which formed GSH conjugates is concluded to be the most likely mechanism for its hepatotoxicity [159].
3.8.2 Transporters and Toxicity
There are no reports on the interaction of Sitaxentan with uptake or efflux transporters.
3.8.3 Risk Factors
The major risk factors associated with Sitaxentan are likely to be environmental and genetic factors that would lead to enhanced formation of the toxic reactive metabolites (e.g. elevation of CYP3A4 activity due to exposure to inducers) or compromised detoxification pathways (e.g. decreased cellular GSH contents due to co‐exposure to GSH depleting agents).
3.9 Sorivudine
Sorivudine (SRV; 1‐beta‐D‐arabinofuranosyl‐ E ‐5‐[2‐bromovinyl] uracil) is a synthetic analog of thymidine intended as an antiviral drug for the treatment of varicellar zoster virus [160–162]. The antiviral activity of SRV is a result of its phosphorylation by thymidine kinase followed by incorporation into viral DNA, inhibiting DNA synthesis and thereby viral replication. SRV is also effective as an antiviral agent toward herpes simplex type 1 virus, and Epstein–Barr virus, and has been proposed as a potential treatment of viral outer retinal necrosis [163, 164]. SRV has high oral bioavailability and superior antiviral activity compared to the available antivirals at the time of its introduction [160, 165]. SRV was approved for marketing in Japan in 1993 and was withdrawn within 40 days of marketing due to its association with 23 cases of severe toxicity, leading to 15 deaths. The deaths and severe gastrointestinal toxicity were observed in cancer patients coadministered SRV and 5‐fluorouracil (5‐FU) prodrugs [166, 167]. Because of the severe toxicity associated with SRV, the drug did not receive regulatory approvals in any other countries, including the United States.
3.9.1 Drug Metabolism and Toxicity
Toxicity observed in patients coadministered SRV and 5‐FU prodrugs is a result of unexpected elevation of plasma 5‐FU levels. The toxicity observed included bone marrow damage, intestinal membrane mucosa atrophy, decreases in white blood cells and platelets, diarrhea with bloody flux, and severe anorexia. Both laboratory animal and clinical results confirm that SRV is metabolized by intestinal flora to ( E )‐5‐(2‐bromovinyl)uracil (BVU), a suicide inhibitor of dihydropyrimidine dehydrogenase (DPD), the key enzyme for 5‐FU metabolism and elimination, leading to retardation of 5‐FU metabolic clearance, resulting in its accumulation to toxic levels [166–168] ( Figure 3.1).
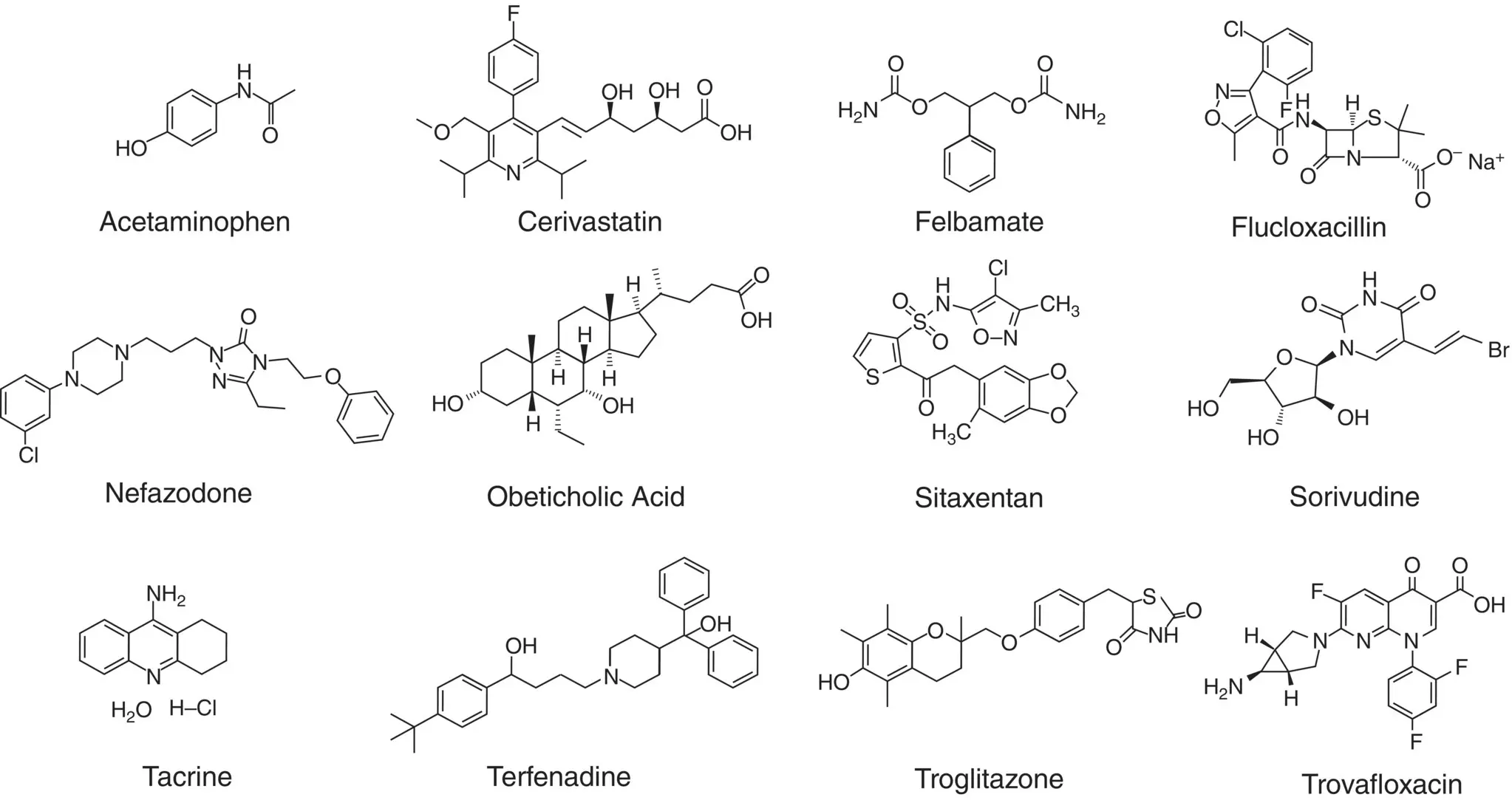
Figure 3.1 Chemical structure of the 12 toxic drugs reviewed.
3.9.2 Transporters and Toxicity
There is no known involvement of transporters in the uptake of SRV.
3.9.3 Risk Factors
Coadministration with 5‐FU is the most important risk factor for the observed lethality.
3.10 Tacrine
Tacrine hydrochloride (1,2,3,4‐tetrahydro‐9‐acridinamine monohydrochloride), an acetylcholinesterase inhibitor [169], was one of the first drugs marketed to combat Alzheimer’s disease based on the cholinergic theory of the disease mechanism. Tacrine was approved for use by US FDA in 1993 to treat Alzheimer’s disease patients with mild to moderate symptoms of dementia. Tacrine was withdrawn from the U.S. market in May 2012 due to liver toxicity and the availability of alternative acetylcholinesterase inhibitor drugs with lower frequency of liver enzyme elevation.
Hepatotoxicity is a major complication of tacrine. It has been estimated that nearly half of the patients that received tacrine had reversible elevation of the clinical marker of liver toxicity, plasma alanine aminotransferase activity. This hallmark of liver toxicity appeared to be reversible in some patients as it was resolved by discontinuation of administration, and in some cases, without discontinuation. However, a small number of patients eventually developed severe and fatal liver toxicity in spite of frequent monitoring of serum liver enzyme activities [170–172].
3.10.1 Drug Metabolism and Toxicity
Toxic metabolite formation: In vitro results suggest that metabolism of tacrine, mainly by CYP1A2 [173], to highly reactive metabolites is a plausible mechanism of its hepatotoxicity. Incubation of tacrine with human liver microsomes has been reported to lead to the formation of mono‐ and dihydroxylated metabolites. Oxidative metabolism to hydroxylated metabolites appeared to involve highly reactive quinone methides that form covalent adducts with macromolecules, leading to hepatotoxicity. This hypothesis was further substantiated by the attenuation of protein binding by tacrine metabolites via supplementation of reduced L‐GSH to human liver microsomes [174–176].
Species differences in metabolism: In vivo [177] and in vitro [178] evaluation of metabolite profiles showed similar metabolite profiles but apparent quantitative differences in tacrine metabolism. A key observation is that human liver microsomes formed the highest levels of reactive metabolites, followed by dog liver microsomes (intermediate) and rat liver microsomes (lowest) [178].
3.10.2 Transporters and Toxicity
As of this writing, there is no evidence that tacrine hepatotoxicity is associated with its interaction with transporters. However, results with rats show that tacrine distribution into rat brains across the blood brain barrier is a function of transporter‐mediated uptake involving organic cation transporters [179].
3.10.3 Risk Factors
High CYP1A2 activities in combination with low GSH levels could be risk factors for tacrine hepatotoxicity. The caffeine breath test, a clinical bioassay for CYP1A2 activity in patients, however, was not successful in the identification of patients with high susceptibility to tacrine hepatotoxicity [180]. On the other hand, there appeared to be correlation between GST polymorphism and susceptibility to tacrine hepatotoxicity [181, 182]. For instance, multivariate Cox hazards model showed that the GST M1‐T1 null genotype was an independent risk factor of tacrine hepatotoxicity [181].
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Transporters and Drug-Metabolizing Enzymes in Drug Toxicity»
Представляем Вашему вниманию похожие книги на «Transporters and Drug-Metabolizing Enzymes in Drug Toxicity» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Transporters and Drug-Metabolizing Enzymes in Drug Toxicity» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.