Biomedical Data Mining for Information Retrieval
Здесь есть возможность читать онлайн «Biomedical Data Mining for Information Retrieval» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Biomedical Data Mining for Information Retrieval
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Biomedical Data Mining for Information Retrieval: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Biomedical Data Mining for Information Retrieval»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Biomedical Data Mining for Information Retrieval — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Biomedical Data Mining for Information Retrieval», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
4 iv) 4-D prediction of the quaternary structure of a multiprotein complex which is made up of more than one peptide chain involving formation of sulfur bridge.
Thus a model development which allows the flexibility of bond formation and helps to predict a stable and functional protein structure has been facilitated to a great deal by AI and ML.
Prediction of protein structure is a complex problem as it is associated with various levels of organization and is a multi-fold process. There is a need for smart computational techniques for such purpose. AI is a great tool which when used with computational biology facilitates such prediction. Apart from determining the structure AI also aids in predicting protein structure crucial for drug development as well as in understanding the biochemical effect and ultimately the function.
A protein can be broadly described as a polymer where the individual amino acid can be considered as the monomers or the building blocks arranged in a linear chain and joined together by peptide bonds. The primary structure as described earlier is represented by a sequence of letters which represent the amino acids. The chain of amino acids of a protein folds into local secondary structures including alpha helices, beta strands, and nonregular coils [35, 36] in its native environment. The secondary structure elements are further packed to form a tertiary structure depending on hydrophobic forces and side chain interactions, such as hydrogen bonding, between amino acids [37–39]. The tertiary structure is described by the x, y and z coordinates of all the atoms of a protein or, in a coarser description, by the coordinates of the backbone atoms ( Figure 2.1). The quaternary structure is formed by more than one protein chains interacting or assembling together to form a complexes structure. Theses protein complexes proteins interact with each other and with other biological macromolecules such as DNA, RNA and certain metabolites in a cell. This kind of interaction is required to carry out various types of biological functions such as enzymatic catalysis (protein complex can interact with a metal or non-metal referred to as co-enzyme), to gene regulation (interaction of transcription factors with DNA sequences), control of growth and differentiation (protein– protein interaction where ligand binding to receptor triggers a signal cascade pathway) and transmission of nerve impulses [40]. A protein’s function is and its structure are dependent on each other [37, 38, 41, 42] therefore, determination or prediction of protein structure accurately holds the key for its function determination. The most effective methods for finding protein structure since the inception of this field have been Nuclear Magnetic Resonance and X-ray crystallography which have the disadvantage of being time consuming and expensive. The recent advancement has been the introduction of cryo-electron microscope (cryo-EM) which produces high-resolution large-scale molecular structures very efficiently. Cryo-EM density maps make use of machine learning and artificial intelligence for prediction [43–46]. For such experiments protein crystal is needed which is the most disadvantageous or complex part of these methods because there are many liquid proteins which do not crystalize. Artificial intelligence comes to our aid here as it is a possible better pathway for sequencing these proteins [47, 48] due to the fact that they have proved their efficacy and accuracy of successful application in different fields like business [49], image recognition to name and can accurately and efficiently predict thousands of possible structures in shortest time by analysing big data where other methods have failed to deliver accurate and useful information.
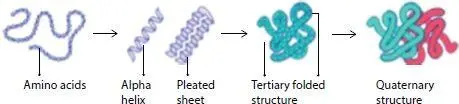
Figure 2.1 The different level of organization of protein.
Most of the models are inaccurate and do not produce predicted proteins that contain useful information so using artificial intelligence, programs are trained using many numerically represented atomic features from the models (such as bond lengths, bond angles, residue-residue interactions, physio-chemical properties, and potential energy properties). Then the comparison of the prediction models output to the known crystal structures helps to assess the quality of the model and find the most accurate model. Models for predictions and prediction analysis are compared each year in one main gathering called the Critical Assessment of Structure Prediction (CASP). Every two years researchers from around the world submit machine learning methods designed for protein structure prediction [50] where the latest advancement has been the help of protein contact distance prediction [51] and addition of quality assessment (QA) category in CASP7 (2006) [51, 52].
AI which is time and resource efficient allows for more accurate prognosis and diagnosis of structures because the computers can analyze data and have perfect calculations and deeply analyze the details. These accuracies while may be very close to that of traditional approaches are still slightly stronger allowing confidence in the results. AI would also help in cost reduction and would not be an agent to replace researchers but rather working in conjunction with them. Artificial Intelligence is an exciting field which offers solutions to issues in finding structures of proteins which is crucial to drug development and the understanding of biochemical effects. A protein’s function is determined by its structure [53–56] as the evidence is there in many biochemical reactions, therefore elucidating a protein’s structure as seen in Table 2.1is key to understanding its function. Function determination in turn is essential for any related biological, biotechnological, medical, or pharmaceutical applications which is much needed in today’s time of increased anti-microbial resistance and threat by unknown biological agents.
Table 2.1 Summary of database sources of protein structure classification.
| Database sources | Websites | References |
|---|---|---|
| PDB | http://www.rcsb.org/pdb/ | [57] |
| UniProt | http://www.uniprot.org/ | [58] |
| DSSP | http://swift.cmbi.ru.nl/gv/dssp/ | [59] |
| SCOP | http://scop.mrc-lmb.cam.ac.uk/ | [60] |
| SCOP2 | http://scop2.mrc-lmb.cam.ac.uk/ | [61] |
| CATH | http://www.cathdb.info/ | [62] |
The various predictive models for protein structure prediction are hidden Markov models, neural networks, support vector machines, Bayesian methods, and clustering methods.
Hidden Markov Model for Prediction HMMs are among the most important techniques for protein fold recognition. In the HMM version of profile–profile methods, the HMM for the query is aligned with the prebuilt HMMs of the template library. This form of profile–profile alignment is also computed using standard dynamic programming methods. Earlier HMM approaches, such as SAM [63] and HMMer [64], built an HMM for a query with its homologous sequences and then used this HMM to score sequences with known structures in the PDB using the Viterbi algorithm, an instance of dynamic programming methods. This can be viewed as a form of profile-sequence alignment. More recently, profile–profile methods have been shown to significantly improve the sensitivity of fold recognition over profile–sequence, or sequence–sequence, methods [65].
Neural Networks (NNs) It is very challenging to determine the structure of a protein if its sequence is given and hence making function determination more difficult. Since a lot of molecular interaction and various levels of folding are involved in a functional protein simple input of sequence will not result in desired output. Deep learning methods are rapidly evolving field in the context of complex relationships between input features and desired outputs which has been put to great use in structure prediction. Various deep neural network architectures resembling the neural network of a human have been proposed which includes deep feed-forward neural networks, recurrent neural networks and neural Turing machines and memory networks. Such advancements are making this field more competitive and accurate and a comparison can be made to a human brain where it receives so many information as inputs but is able to analyze and come to a logical conclusion.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Biomedical Data Mining for Information Retrieval»
Представляем Вашему вниманию похожие книги на «Biomedical Data Mining for Information Retrieval» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Biomedical Data Mining for Information Retrieval» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.